Targeting KRAS in pancreatic adenocarcinoma: Progress in demystifying the holy grail
2023-10-18AhmedElhaririAhmedAlhajDanielAhnMohamadBassamSonbolTaniosBekaiiSaabChristinaWuMichaelScottRutenbergJohnStaufferJasonStarrUmairMajeedJeremyJonesMiteshBoradHaniBabiker
Ahmed Elhariri,Ahmed Alhaj,Daniel Ahn,Mohamad Bassam Sonbol,Tanios Bekaii-Saab,Christina Wu,Michael Scott Rutenberg,John Stauffer,Jason Starr,Umair Majeed,Jeremy Jones,Mitesh Borad,Hani Babiker
Abstract Pancreatic cancer (PC) remains one of the most challenging diseases,with a very poor 5-year overall survival of around 11.5%.Kirsten rat sarcoma virus (KRAS)mutation is seen in 90%-95% of PC patients and plays an important role in cancer cell proliferation,differentiation,metabolism,and survival,making it an essential mutation for targeted therapy.Despite extensive efforts in studying this oncogene,there has been little success in finding a drug to target this pathway,labelling it for decades as “undruggable”.In this article we summarize some of the efforts made to target the KRAS pathway in PC,discuss the challenges,and shed light on promising clinical trials.
Key Words: Kirsten rat sarcoma virus;Targeted therapy;Pancreatic cancer;Drug resistance;Next generation sequencing;Clustered regularly interspaced short palindromic repeats
INTRODUCTION
In 2022,there was an estimated 62210 new pancreatic cancer (PC) cases and 49830 estimated deaths.PC is the fourth leading cause of cancer death in the United States[1].PC is driven primarily by mutations in the Kirsten rat sarcoma virus(KRAS) gene,cyclin-dependent kinase inhibitor 2A,tumor protein 53,and mothers against decapentaplegic protein homolog 4.KRASis one of the most frequently mutated oncogenes in human cancers.It is seen in more than 90% of PCs and more than 40% of colorectal and lung cancers[2].93% of allKRASmutations occur at codon 12 (G12) with other common mutation sites at G13 and Q61.Missense mutation in glycine residues of G12 result in amino acid substitution,glycine substituted with aspartic acid (G12D),with valine (G12V),or with cysteine (G12C)[3].The predominant mutation in PC is G12D followed by G12V (Figure 1)[4],but in lung cancer G12C is the most common.KRASplays a major role in the development of PC and,as a result,there have been significant efforts to target the mutatedKRASpathway.
BACKGROUND
KRASis a member of the rat sarcoma viral oncogene family (RAS),in addition to Neuroblastoma rat sarcoma virus and Harvey rat sarcoma virus.Identified in 1982,theKRASis located on the short arm of chromosome 12[5,6].It encodes two protein isoforms,KRAS-4B and KRAS-4A.Those are found in the inner side of the plasma membrane[7],and act as guanosine triphosphate (GTP)-binding proteins (G proteins),they bind guanine nucleotides that belong to the family of GTP-bound regulatory protein phosphatases (GTPase).An upstream signale.g.,epidermal growth factor receptor (EGFR)stimulates the dissociation of guanosine diphosphate (GDP) from the GDP-bound G protein form,and allows the binding of GTP[8].RAS functions as a binary switch,determined by two regulatory proteins called guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAP)[9].KRAS binds to GDP in resting state due to its intrinsic GTPase activity.But with relevant stimuli,GEFs turn on signaling by catalyzing the exchange from a KRAS G-protein-bound GDP to GTP[10] (Figure 2).KRAS proteins can be activated by tyrosine kinase receptors,growth factors,chemokines,or calcium.This in turn activates multiple signaling pathways including the rapidly accelerated fibrosarcoma (RAF)-mitogen-activated protein kinase (MAPK)-extracellular regulated protein kinases (ERK) (MAPK/ERK;MEK) signaling pathway,the phosphoinositide 3-kinase (PI3K)-protein kinase (AKT)-mammalian target of rapamycin (mTOR) signaling pathway,and others.These pathways result in cell proliferation and DNA synthesis (Figure 3).
Precursor lesions of pancreatic ductal adenocarcinoma (PDAC) include pancreatic intraepithelial neoplasia (PanIN),intraductal papillary mucinous neoplasm (IPMN),and mucinous cystic neoplasm[11,12].KRASmutation was detected in 36% of PanIN-1A lesions and 87% of PanIN-2-3 lesions[13].It was also found in 61% of patients with IPMN[14].To study the role ofKRASin PC progression,scientists developed transgenic mice with inducibleKRASG12D.Induction of oncogenicKRASG12Daltered normal epithelium and led to the development of precancerous lesions;on the other hand,inactivation ofKRASG12Din precursor lesions and during cancer progression led to disease regression[15].These studies confirm the early role ofKRASmutation in the initiation and progression of precursor lesions into invasive PDAC as well as the correlation between frequency ofKRASmutation and degree of dysplasia.
KRASmutation drives PC progression by resistance to apoptosis,induction of autophagy[16],immune evasion by downregulating major histocompatibility complex class I on tumor cells[17],and stimulating angiogenesis,resulting in cell survival and tumor progression.
TARGETED THERAPY
Upstream regulators
Some of the key regulators of KRAS include Son of Sevenless (SOS) and Src homology phosphatase 2 (SHP2).SOS is a GEF that activates KRAS,and SHP2 is a protein tyrosine phosphatase encoded byPTPN11that also promotes RAS activation,inhibiting either can delay tumor progression[18,19].
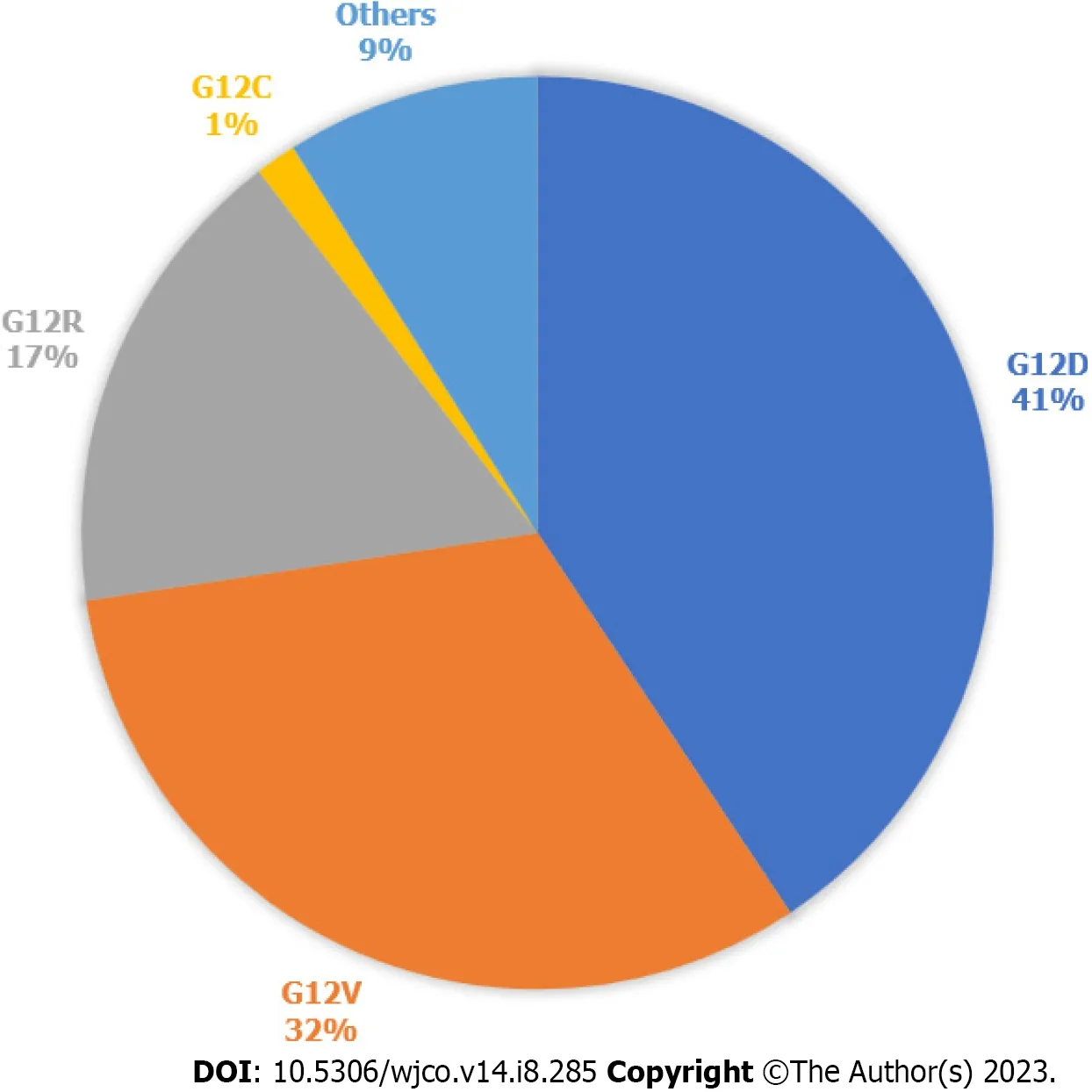
Figure 1 Kirsten rat sarcoma virus mutations in pancreatic cancer. Types of Kirsten rat sarcoma virus (KRAS) mutations seen in pancreatic cancer,according to data publicly available on cBioPortal.812 samples with altered KRAS collected from 5 pancreatic cancer studies.Others are A11T,A146T,A18V,G12A,G12I,G12L,G12S,G13C,G13D,G13H,G13P,G13R,L23V,Q61H,Q61K,Q61R.
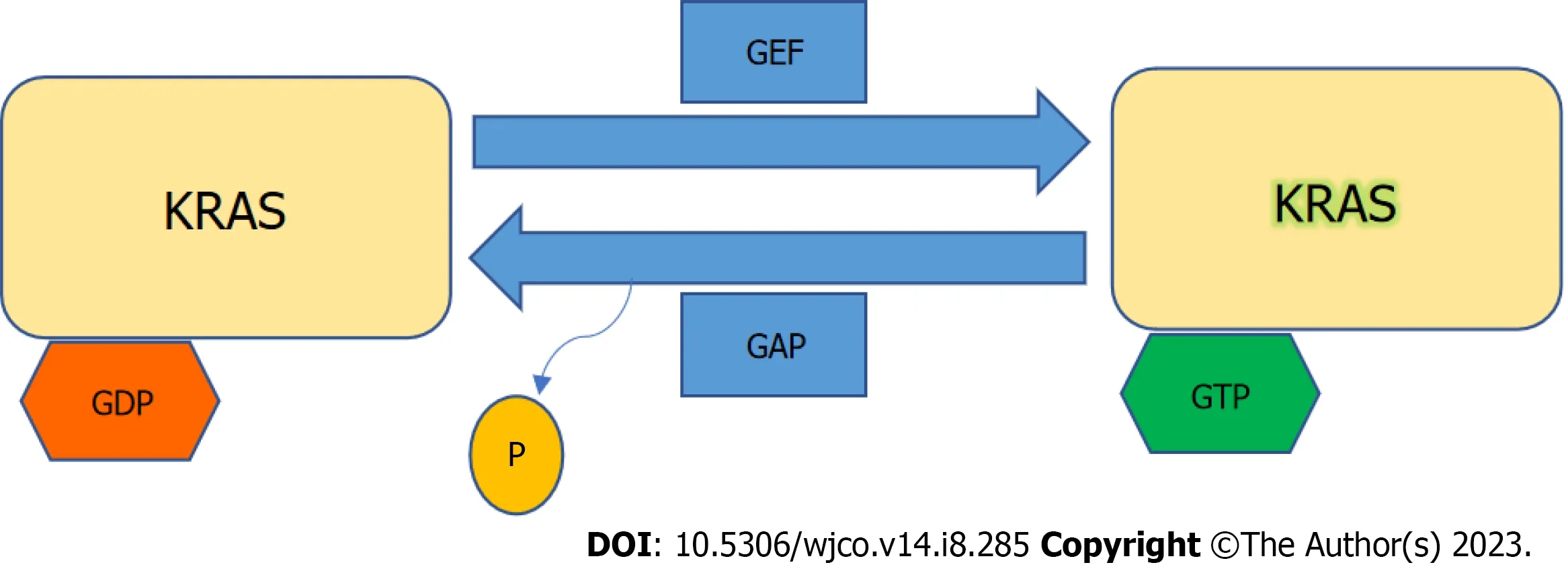
Figure 2 Kirsten rat sarcoma virus activation. Kirsten rat sarcoma virus is activated when guanine nucleotide exchange factor displaces guanosine diphosphate from nucleotide binding site allowing guanosine triphosphate (GTP) binding and inactivated upon GTP hydrolysis by intrinsic GTP-bound regulatory protein phosphatases (GTPase) activity enhanced by GTPase activating protein.GTP: Guanosine triphosphate;GAP: GTPase activating protein;GDP: Guanosine diphosphate;GEF: Guanine nucleotide exchange factor;KRAS: Kirsten rat sarcoma virus.
BI-3406 inhibits the interaction between KRAS and SOS1 which has been shown to cause tumor regression in KRASdriven cancer cell models.Synergy was observed with SOS1/MEK inhibitors as this combination can counteract adaptive resistance to MEK inhibitors[20].ERAS-601 is a small molecule allosteric inhibitor of SHP2 that stops KRAS from cycling into its GTP-active state,which inhibits cellular proliferation in multipleKRASG12Cmutated tumor cell models[21].Recently the Food and Drug Administration (FDA) granted fast track designation to BBP-398 (SHP2 inhibitor) in combination with Sotorasib forKRASG12C-mutated metastatic non-small-cell lung carcinoma (NSCLC).There is an ongoing trial to evaluate the safety and efficacy of this combination [national clinical trial (NCT) 05480865].Combination of KRASG12Cinhibitor (JAB-21822) and SHP2 inhibitor (JAB-3312) showed synergistic effect inKRASG12C-resistant tumor cells[22],currently in phase I/II trial for PDAC (NCT05288205).
MAPK/ERK pathway
The MAPK/ERK pathway was shown in Table 1.
KRAS
Direct inhibition of the KRAS protein remains a challenge,due to its small size of 21 kDa and the lack of hydrophobic pockets on its surface.Those pockets,if found,can then be blocked by small molecules and ultimately disrupt its interaction with other proteins[23].Several attempts have been made to directly target KRAS,but results were non-satisfactory[24-26].Only recently AMG 510 (sotorasib) was developed to target G12C mutation in NSCLC without inhibiting wild-typeKRAS[27].Adagrasib (MRTX849) which is also a KRASG12Cinhibitor is well tolerated,and preliminary results showed partial response in 50% of patients with PDAC harboring this mutation[28].However,KRASG12Conly occurs in 1%-2% PC and attempts to target more commonKRASisoforms have failed.One promising compound is MRTX1133,a small molecule that selectively inhibits KRASG12Dby preventing SOS-catalyzed nucleotide exchange.Subsequently,it promotes tumor regression in immunocompetent PC models and alters the tumor microenvironment by increasing tumor associated macrophages (TAM) and tumor-infiltrating cytotoxic T-cells.MRTX1133 is expected to enter phase I trial in 2023[29,30].Other agents inhibiting G12D in the pre-clinical phase include BIKRASG12D,JAB-22000,and ERAS-4.A new category of drugs called tricomplex inhibitors has shown promising results in pre-clinical models ofKRASG12Vmutant cancers[31] and in a phase I trial RMC-6236 inKRASG12-mutant advanced solid tumors excluding G12C (NCT05379985).A recent study was able to selectively target KRASG12Rusing a small molecule electrophile[32].Due to the challenging nature of direct KRAS inhibition focus was shifted on downstream signaling,knowing that some of the challenges include compensation by other pathways,and that inhibiting multiple pathways can result in toxicity[33].
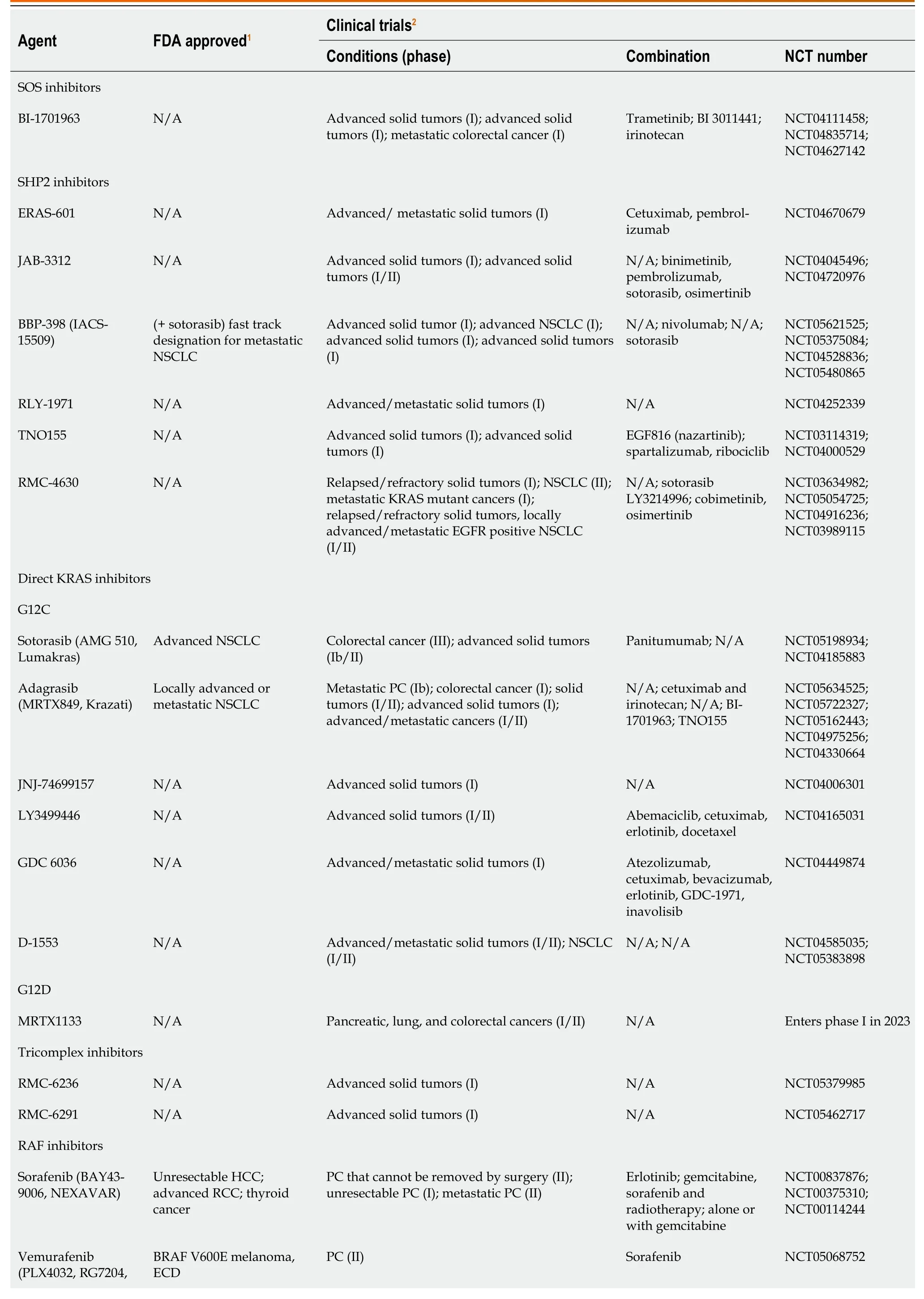
Table 1 Kirsten rat sarcoma virus-rapidly accelerated fibrosarcoma-mitogen-activated protein kinase/extracellular regulated protein kinases-extracellular regulated protein kinases pathway inhibitors
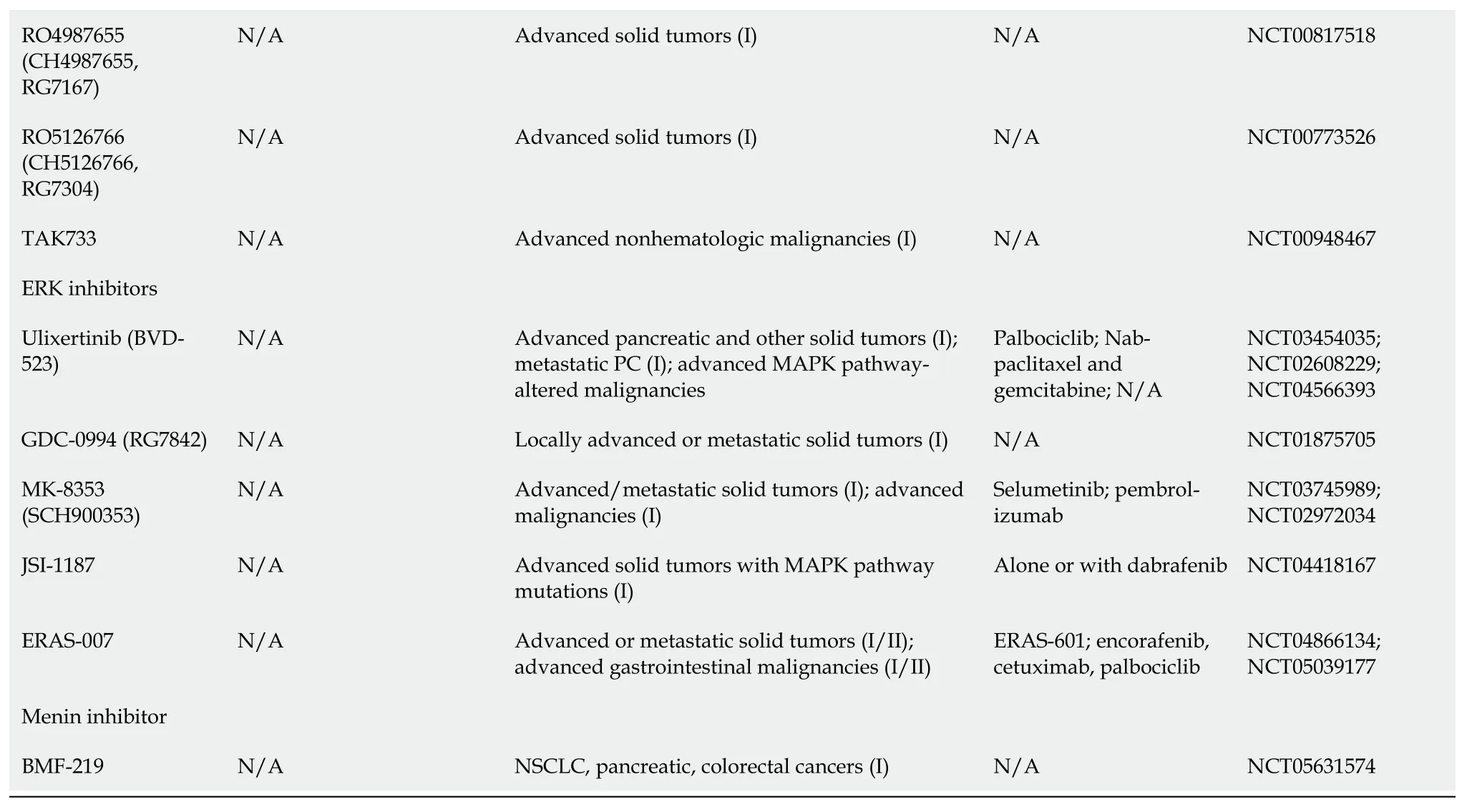
1www.fda.gov.2clinicaltrials.gov.FDA: Food and Drug Administration;SOS: Son of Sevenless;KRAS: Kirsten rat sarcoma virus;HCC: Hepatocellular carcinoma;RCC: Renal cell carcinoma;ECD: Erdheim-Chester disease;GIST: Gastrointestinal stroma tumors;PC: Pancreatic cancer;RAF: Rapidly accelerated fibrosarcoma;RAS: Rat sarcoma viral oncogene family;MAPK: Mitogen-activated protein kinases;NSCLC: Non-small-cell lung carcinoma;SHP2: Src homology-2 domain-containing protein tyrosine phosphatase-2;NCT: National clinical trial;MEK: Mitogen-activated protein kinase/extracellular regulated protein kinases;N/A: Not applicable.
Multiple mechanisms are implicated in the inevitable drug resistance seen with KRAS inhibitors,either by activation of wild-type KRAS which is mediated by receptor tyrosine kinase[34],synthesizing new KRASG12Cproteins in response to MAPK suppression[35],or developing secondary mutations in KRAS inhibitor binding pocket[36].
RAF
With regards to drugs targeting the MAPK pathway,sorafenib was the first RAF inhibitor to be FDA-approved,initially for advanced renal cell carcinoma,followed by unresectable/metastatic hepatocellular carcinoma and metastatic differentiated thyroid cancer[37].In a phase II trial combining sorafenib and erlotinib,12 of the first 15 patients required dose delays or reductions due to toxicity,and the study failed to reach its primary endpoint of 8-week progression-free survival (PFS)[38].A second-generation of RAF inhibitors (e.g.,vemurafenib and dabrafenib) was proven to be effective inBRAFV600Emutant metastatic melanoma[39].Dabrafenib in combination with trametinib received a tumor agnostic accelerated approval for treatment of unresectable/metastatic solid tumors withBRAFV600Emutation that progressed on prior treatment[40].Unfortunately,vemurafenib and dabrafenib were not as effective inKRAS-mutant cancers,due to compensatory ERK activation that led to enhanced tumor growth[41,42].A third generation of RAF inhibitors called“paradox breakers” (PLX7904 and PLX8394) also blocks MEK-ERK1/2,which can overcome this resistance mechanism[43],Unfortunately,a phase I/II trial to evaluate the safety of PLX8394 was terminated due to low accrual.Recently,another group called “pan-RAF inhibitors” (LY3009120,MLN2480,and HM95573) entered phase I trials.LY3009120 is a kinase inhibitor that showed efficacy in inhibiting mutatedKRASandBRAFin preclinical models of colorectal cancer with minimal paradoxical MAPK activation[44,45],however,a phase I trial in advanced cancers was terminated early due to lack of sufficient clinical efficacy (NCT02014116).MLN2480 (tovorafenib) showed an acceptable safety profile[46],and HM95573 (belvarafenib) was well tolerated and showed anti-tumor activity in advanced solid tumors with RAS or RAF mutations[47].The Yes-associated protein (YAP) is a transcription coregulator downstream from KRAS that promotes cell proliferation[48].Combining LY3009120 and YAP-inhibitor (verteporfin) showed anti-tumor effectinvivoandinvitroby blocking compensatory activation of AKT pathway[49].
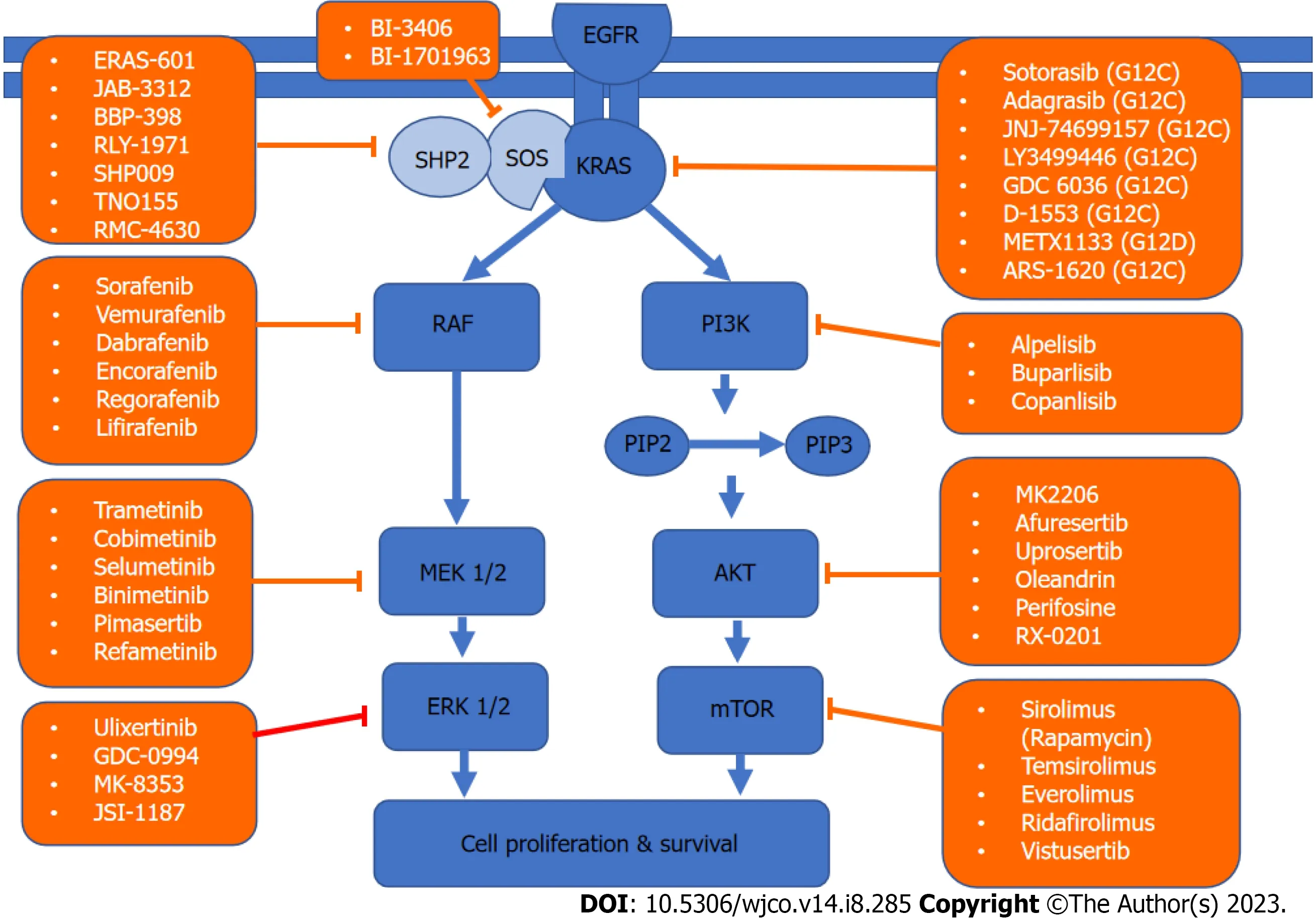
Figure 3 Kirsten rat sarcoma virus signaling network and targeted therapy. A schematic of the two major Kirsten rat sarcoma virus pathways driving cell survival and drugs that target them.KRAS: Kirsten rat sarcoma virus;AKT: Protein kinase;EGFR: Epidermal growth factor receptor;PIP: Prolactin-induced protein;ERK: Extracellular regulated protein kinases;MEK: Mitogen-activated protein kinase/extracellular regulated protein kinases;mTOR: Mammalian target of rapamycin;PI3K: Phosphoinositide 3-kinase;RAF: Rapidly accelerated fibrosarcoma;SHP2: Src homology-2 domain-containing protein tyrosine phosphatase-2;SOS: Son of sevenless.
MEK
As mentioned above,trametinib is a MEK1/2 inhibitor FDA approved in combination with dabrafenib (RAF-inhibitor) as a tumor agnostic drug[50].Trametinib was studied in combination with gemcitabine in a placebo controlled clinical trial for untreated metastatic PDAC.Unfortunately,it did not show improvement in overall survival (OS),PFS,or overall response rate (ORR)[51].This is potentially due to a compensatory mechanism called autophagy,initiated through activation of the AKT pathway[52].A Phase II trial of selumetinib (MEK1/2 kinase inhibitor) in PC did not show any significant difference in OS when compared to capecitabine[53],another phase II study of selumetinib targeting only PC patients withKRASG12Rmutation after at least two lines of prior systemic chemotherapy did not improve ORR,however,three patients had stable disease for ≥ 6 months[54].A phase I/II trial studied the selective MEK1/2 inhibitor pimasertib in combination with gemcitabinevsgemcitabine alone in patients with metastatic PC.Despite the promising safety and efficacy of this combination,it did not improve PFS or OS[55].Unfortunately,in whole there was no observed clinical benefit of MEK inhibitors in the multiple trials done in PC.
ERK
After resistance to BRAF and MEK inhibitors,the next downstream target is ERK.SCH772984[56] is a selective inhibitor of ERK1/2 that showed tumor regression in xenograft models refractory to BRAF and MEK inhibitors.Similar effects were seen with ulixertinib[57].A phase Ib trial combining ERK1/2 inhibitor (GDC-0994) and MEK inhibitor (cobimetinib)in advanced solid tumors was terminated due to tolerability issues[58].The ERK1/2 inhibitor JSI-1187-01 demonstrated pre-clinical efficacy in tumor models with MAPK pathway mutations,as well as synergy with BRAF inhibitors[59],and is being studied in a phase I trial (NCT04418167).
PI3K-AKT-mTOR-pathway
The PI3K-AKT-mTOR-pathway was shown in Table 2.One of the postulated reasons EGFR inhibitors and other targeted therapies develop resistance is the hyper-activation of PI3K-AKT-mTOR pathway,which can drive cancer progression and survival.PI3K is overexpressed in around 50% of patients with PC[60],and AKT2 is amplified in 10%-20% of PDAC[61].TAM plays a role in the development of PC[62] by creating an immune-suppressive microenvironment,minimizingthe antitumor effect of T-cells[63].PI3K helps drive this immune suppression,so its inhibition can restore immune response against cancer cells as well as potentiate the effect of chemotherapy[64].Additionally,AKT mediates an antiapoptotic effect and plays a role in chemoresistance[65].Phosphatase and tensin homolog is a tumor suppressor of the AKT/mTOR pathway,its loss has been implicated in PC development,recurrence,and prognosis[66],as well as acceleration ofKRASG12D-induced PDAC in mice[67].Aninvivostudy tested PI3Kα-specific inhibitor (BYL) in combination with an EGFR inhibitor (erlotinib) and showed reduced tumor volume and apoptosis in PDAC cell lines[68].Currently a clinical trial combining gedatolisib (PI3K/mTOR inhibitor) with palbociclib (CDK4/6 inhibitor) in advanced squamous cell cancers of the lung,pancreas,and solid tumors is recruiting (NCT03065062).A phase I/II trial studied the safety and efficacy of combining everolimus (mTOR inhibitor),cetuximab (EGFR inhibitor),and capecitabine,however,the combination resulted in significant epidermal and mucosal toxicities with minimal efficacy[69].
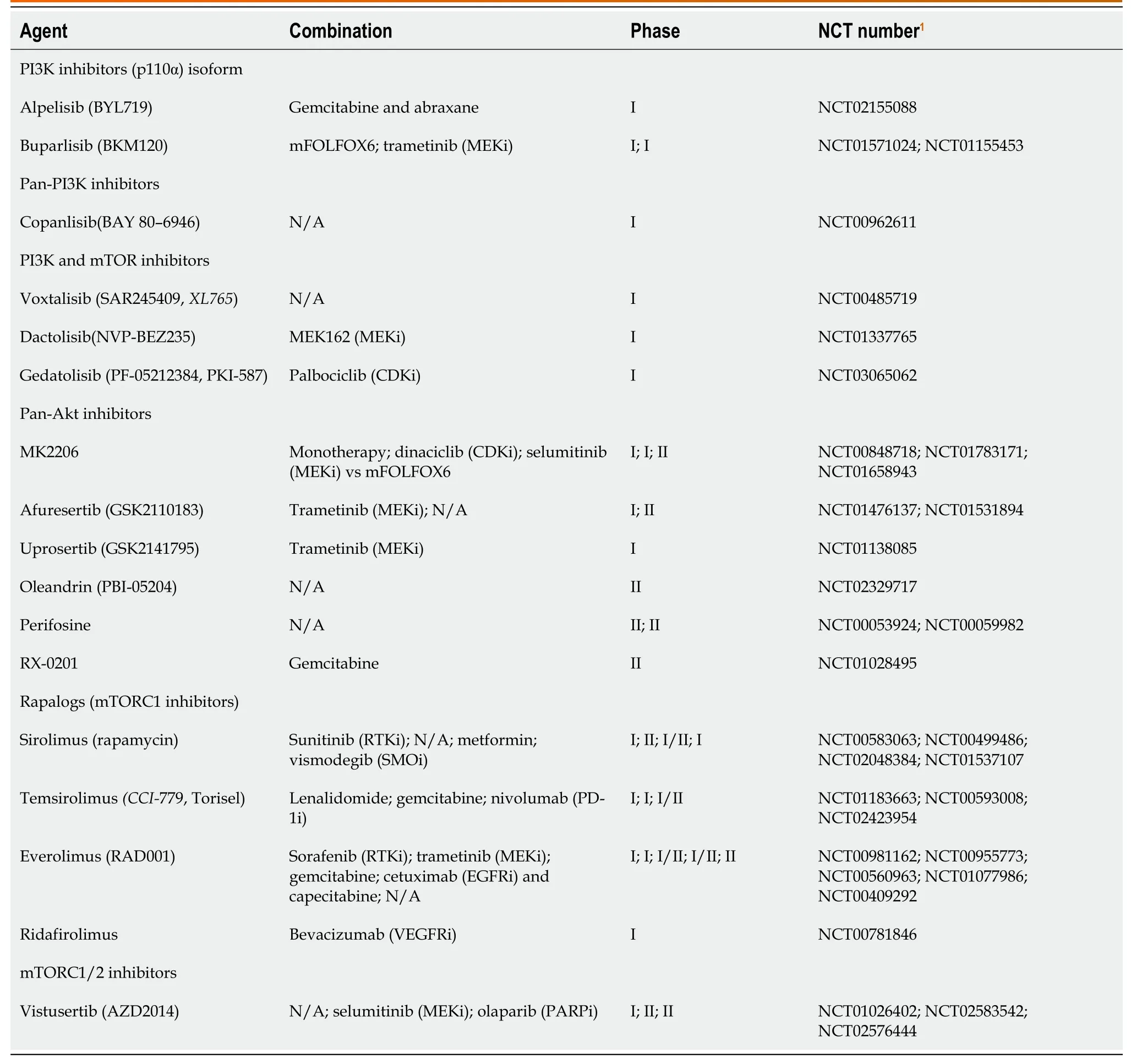
Table 2 Phosphoinositide 3-kinase-protein kinase-mammalian target of rapamycin-pathway inhibitors
Small interfering RNA,MicroRNA,and clustered regularly interspaced short palindromic repeats
Pre-clinical studies show that small interfering RNAs (siRNAs) have potential in cancer treatment.To deliver siRNAs to target cancer cells,scientists devised two unique methods,one utilized nanoparticle[70] to target lung cancer cells and another study used a biodegradable polymeric matrix (LODER) to carry the antiKRASG12DsiRNA.This resulted in the decrease of KRAS levels and inhibited cell proliferation[71].MicroRNAs (miRNA) regulate cell proliferation and contribute to PC development.Depending on their role they can act as tumour suppressor or oncogenic miRNAs[72,73].MRX34 (miRNA-34 mimic) was used in a phase I clinical trial that utilized lipid-based vesicles (NOV40) as a delivery vector,for treating patients with advanced solid tumors.miRNA-96 directly targetsKRASoncogene decreasing PC cell invasion and slowing tumor growth bothinvivoandinvitro[74].Clustered regularly interspaced short palindromic repeat (CRISPR) is currently being studied inKRAS-mutated cancers.This technology is being harnessed to target inactivated tumor suppressor genes or overactive oncogenes.In a 2018 study CRISPR-Cas13a was developed to targetKRASG12DmRNA.Subsequently,it also suppressed downstream ERK and AKT proteins resulting in apoptosis and significant tumor suppressioninvivoandinvitro[75].Two phase I trials utilizing the CRISPR platform are currently ongoing in PC (NCT04426669 and NCT04842812).
CONCLUSION
KRASmutation remains the hallmark genetic aberration leading to PC.Although several studies have demonstrated positive preclinical results,the resulting clinical trial results have been largely disappointing.As we continue to have a deeper understanding of the KRAS pathway,resistance mechanisms,and the role and function of the immune system;we get closer to developing effective therapies to outsmart the scourge that is PC.Ongoing clinical trials targeting more commonKRASmutations in PC will hopefully lead to more effective therapy and change the outcomes for the thousands of patients affected by this disease every year.
ACKNOWLEDGEMENTS
Dr.Babiker is a Paul Calabresi Scholar at the Mayo Clinic Cancer Center and acknowledges K-12 grant Program,K12CA090628.
FOOTNOTES
Author contributions:Elhariri A and Babiker H designed and wrote the manuscript;Alhaj A,Ahn D,Sonbol MB,Bekaii-Saab T,Wu C,Rutenberg MS,Stauffer J,Starr J,Majeed U,Jones J and Borad M critically reviewed and edited the manuscript;All authors approved the final version of the manuscript.
Conflict-of-interest statement:Authors declare no conflict of interests for this article.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BY-NC 4.0) license,which permits others to distribute,remix,adapt,build upon this work non-commercially,and license their derivative works on different terms,provided the original work is properly cited and the use is non-commercial.See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:United States
ORCID number:Ahmed Elhariri 0000-0002-3715-4842.
S-Editor:Lin C
L-Editor:A
P-Editor:Xu ZH
