马铃薯与致病疫霉互作研究进展与展望
2023-10-16田峰奇王路遥董莎萌
田峰奇, 王路遥,2, 董莎萌*
(1. 南京农业大学植物保护学院,卵菌与真菌分子生物学实验室,南京 210095;2. 中国农业科学院深圳农业基因组研究所,深圳 518120)
马铃薯是关涉我国粮食安全和脱贫攻坚重大战略的农作物之一,兼具主粮、菜用和饲用等重要功能。我国于2009年启动马铃薯产业技术体系建设,依托技术研发中心和各地试验站,推动马铃薯育种、病虫害防控、土肥栽培、机械、贮藏加工、产业经济等相关领域的研究[1]。2013年,国家马铃薯主粮化项目组成立并着手开展马铃薯主粮化关键技术研发与示范工作,初步验证了马铃薯主粮化的可行性[2]。2015年1月,中国农业科学院、国家食物与营养咨询委员会、中国种子协会共同召开马铃薯主粮化发展战略研讨会,会议强调了推进马铃薯主粮化战略的必要性,提出了马铃薯主粮化的目标和基本原则,标志着我国马铃薯主粮化战略正式启动[3-4];2015年6月,农业部办公厅印发《农业部贯彻落实党中央国务院有关“三农”重点工作实施方案》,将马铃薯主粮化战略作为工作重点[5]。随着主粮化战略的推进,我国马铃薯相关产业发展迅速,2021年栽培面积达578万hm2(FAO数据)[6],目前已成为全世界马铃薯种植面积最大的国家,且马铃薯消费潜力仍未得到充分挖掘[7]。保障我国马铃薯安全生产已成为当前确保我国粮食安全的重要路径之一。
作物疫病是全球农业生产中的重大病害之一,给全球的粮食和生态安全带来严重威胁,每年造成我国马铃薯、番茄等蔬菜作物及大豆等经济作物的经济损失高达数百亿元[8]。其中,晚疫病(late blight)是由致病疫霉Phytophthorainfestans侵染马铃薯造成的毁灭性病害,也是目前马铃薯产业发展的重要障碍之一,因其历史上直接引发了19世纪40年代的爱尔兰大饥馑而备受关注,100多年来,科学家持续不断地与迅速变异的晚疫病菌作斗争。目前,我国大量栽培中感、高感晚疫病的马铃薯品种,感病品种种植面积达50%,受晚疫病威胁大[9-12]。近年来,我国晚疫病发生范围广、危害大,年平均发病面积近200万hm2,可达马铃薯平均种植面积的40%;2012年,我国北方马铃薯产区6月-8月降水量高于往年同期,较高的湿度为晚疫病发生创造了条件,全国多个产区发生晚疫病大流行,甘肃、湖北等省份晚疫病发生面积超过种植面积的80%,病株率一般60%~100%,部分地区甚至绝收;2008年-2017年全国马铃薯晚疫病年平均造成损失37.67万t,占所有马铃薯病害造成损失的63.54%;2017年-2021年间,云南红河州东马铃薯产区晚疫病发病面积占种植面积的近30%,使当地马铃薯产业严重受损[13-15]。2023年,预测我国马铃薯晚疫病发病面积可达166.67 hm2[12],为晚疫病防治提出了高要求。晚疫病也同样威胁全球马铃薯生产,例如爱沙尼亚持续受到多变的晚疫病菌株的威胁,2012年当地晚疫病发生较重,一些地区感病品种的马铃薯叶片在12~15 d内被完全破坏[16-17];在印度,马铃薯和番茄晚疫病每隔2~3年会发生大流行,造成高达75%的产量损失[18-19]。Savary等调查2010年-2014年全球作物生产情况,分析病虫害对主要作物造成的产量损失,发现病虫害引起全球马铃薯产量损失约17.2%,其主要损失来自马铃薯晚疫病,在全球引起损失超过7%,个别地区超过10%,远超其他马铃薯病虫害[20]。
欧洲于2009年启动了EuroBlight项目,该项目前身来自1996年-2000年的欧洲马铃薯晚疫病综合防治战略发展网络(European network for development of an integrated control strategy of potato late blight),以及2003年-2006年的欧洲马铃薯晚疫病网络(a potato late blight network for Europe),旨在统合晚疫病的监测和研究结果,开展全面、高效的晚疫病防控[21]。2022年,EuroBlight收集了来自22个国家和地区的1 100余份致病疫霉菌株,发现含有EU43基因型的晚疫病样本比例正在增加,而这一菌株已被证实对双炔酰菌胺(mandipropamid)等杀菌剂表现出抗药性[22],这为后续晚疫病的防治策略提供了重要参考。与之类似,美国于2011年启动了USABlight项目,开展晚疫病的监测与分析工作[23]。我国农业农村部2023年颁布的《一类农作物病虫害名录》中包含10种虫害和8种病害,马铃薯晚疫病位列其中[24],明确了晚疫病防治为我国重要的植保工作之一。近年来,我国开展了一系列马铃薯抗晚疫病育种专项研究,同时国家重点研发计划也对相关研究起到了重要推动作用[25-26];农业农村部多次公布马铃薯晚疫病防控技术方案、马铃薯重大病虫害防控技术方案、粮食作物重大病虫害防控技术方案,建立防控体系,为我国晚疫病防治工作提供保障[27-29]。在这样的背景下,进一步解析马铃薯晚疫病致病机制和分子互作机理,为推动晚疫病防治提供理论基础,已成为当下植物病理学研究的重要目标。本文主要综述致病疫霉致病机制和寄主抗病机理领域的研究进展,以期对未来研究和防治晚疫病提供参考。
1 我国晚疫病研究状况概要
我国对马铃薯晚疫病的研究可追溯到20世纪中叶。1952年,杨昌寿等报道重庆近郊的巴县新发乡连年受晚疫病为害,造成产量损失超过80%,并通过建立示范田、宣传预防性喷施波尔多液,取得了一定的防治效果;杨昌寿等明确提出,彻底解决马铃薯晚疫病需要选育良种、合理栽培等多种手段结合[30]。关益周报道山西省雁北地区马铃薯晚疫病危害严重,部分田块引发绝收,对抗性品种需求迫切,并在田间通过改良栽培措施提高晚疫病抗性[31]。1953年-1954年,林传光等在河北省张家口地区的农业试验站开展了晚疫病田间观察与试验,其结果于1955年发表于《植物病理学报》,这是我国第一篇系统性研究晚疫病致病规律和防治方法的研究论文,对晚疫病发病时间、中心病株特征、每一代传播距离、病害蔓延速度、平均病程时长等特征进行了观测与描述,并检验了铜离子杀菌剂的防治效果,测试了最优的施用方法[32]。之后,林传光等继续开展晚疫病田间研究,通过接菌试验和田间观察,认为当地马铃薯晚疫病主要初侵染源是带菌薯块;对中心病株的产生和病原菌传播规律进行了分析和解读,强调气象环境对晚疫病发病情况影响极大,可以根据天气情况预测中心病株的出现时间;并提议在各地开展试验,记录晚疫病发展情况,为预测预报提供支持[33-35]。此后,全国各地展开了对马铃薯晚疫病预测预报和防治方法的研究。有机汞制剂、植物源农药开始被应用于晚疫病防治,改变了当时完全依赖铜离子制剂的局面[36-37];新栽培措施得到推广,例如湖北宣恩县劳动模范徐树民开发的催苗方法和配套栽培措施有助于马铃薯苗提前出土、更早成熟,减轻晚疫病的危害[38];同期,对不同品种的抗病性评估和抗性育种研究逐渐展开,筛选出一系列对晚疫病抗性显著的马铃薯品种[39-40]。1999年,四川省农业科学院何卫博士和河北农业大学张志铭教授应邀在全球马铃薯晚疫病防治行动组织(Global Initiative on Late Blight,GILB)开展的世界马铃薯晚疫病学术大会上发言,介绍我国晚疫病综合防治研究成果[41]。近年来,我国研究人员在相关应用领域内的研究不断获得突破,对现有抗病资源的解析和利用为新抗病资源的发掘带来了一系列抗病基因储备[42-43],新型生防制剂、生物源农药和新型RNA农药表现出良好的防效,有助于进一步减少化学农药的施用量[44-46];基于物联网技术的晚疫病监测手段结合预测预报技术,能够有效预测晚疫病流行情况[47]。各方面应用技术的发展一定程度上有效地指导晚疫病的田间防治。
我国在致病疫霉生物学特性和病理学基础理论层面也取得重要进展。1962年,张明厚等注意到,分离自不同抗性水平马铃薯品种上的致病疫霉菌株致病性存在差异,抗病品种上分离的菌株可以侵染感病品种,而感病品种分离菌株则难以侵染抗病品种[48]。周茂繁也记录了类似的现象,且证明感病品种分离的弱毒力菌株在中抗品种上连续多代接种、适应后,致病性增强,可以侵染抗病品种。尽管受限于理论局限,但基于这些现象,研究者已意识到监测致病疫霉生理小种演变的重要性[49]。在这之后,越来越多研究针对马铃薯晚疫病发病特点、致病机制、抗性品种选育、化学防治手段和预测预报方法开展,为我国马铃薯晚疫病防治提供了扎实的理论基础[50-53]。研究者于2003年和2010年分别对我国多地的致病疫霉菌株进行了整理和分析,归纳了生理小种分布,并鉴定到能克服大量抗病基因的超级毒力小种[54-55];2019年,又对我国马铃薯病虫害发生情况和防治现状进行总结[56]。2002年,Ballvora 等克隆了马铃薯抗病基因R1,这是第一个被成功克隆的抗晚疫病基因[57]。随后,我国学者黄三文克隆到马铃薯抗晚疫病基因R3a,并参与筛选与R3a相对应的马铃薯晚疫病无毒基因Avr3a[58-60],验证了马铃薯-晚疫病菌互作过程中的基因对基因学说。近年来,我国科研人员在晚疫病菌与病原物互作机制领域取得了许多成果,例如董莎萌教授课题组发现致病疫霉效应分子调控寄主选择性剪接的新机制[61];詹家绥教授课题组发现致病疫霉调控寄主气孔张开的新毒力机制以及效应分子逃避寄主免疫的新原理[62-63];田振东教授课题组鉴定了一系列在马铃薯-晚疫病菌互作过程中发挥关键作用的信号分子[64-66],大大丰富了基础理论。相关理论的迅速发展将为晚疫病的防治提供新的思路。
2 致病疫霉侵染机制研究进展
2.1 致病疫霉的侵染与致病过程
致病疫霉具有发达的无隔菌丝,可以在寄主植物或植物残体表面生长,并形成不同类型的孢子用于扩散或抵抗不良环境。一部分菌丝顶端会生长出多核的孢子囊(sporangium)[67],在较高的温度下,孢子囊可以直接萌发,从孢子囊壁上产生新生菌丝;而在低温、潮湿的环境下,孢子囊将会发生细胞质分裂,形成游动孢子(zoospore)并将其释放,这一诱导过程被称为“冷休克”(cold shock);游动孢子活动一段时间后休止,并萌发出芽管,侵染寄主植物(图1,图2)。孢子囊和游动孢子都不能耐受长时间的恶劣环境[67-68]。
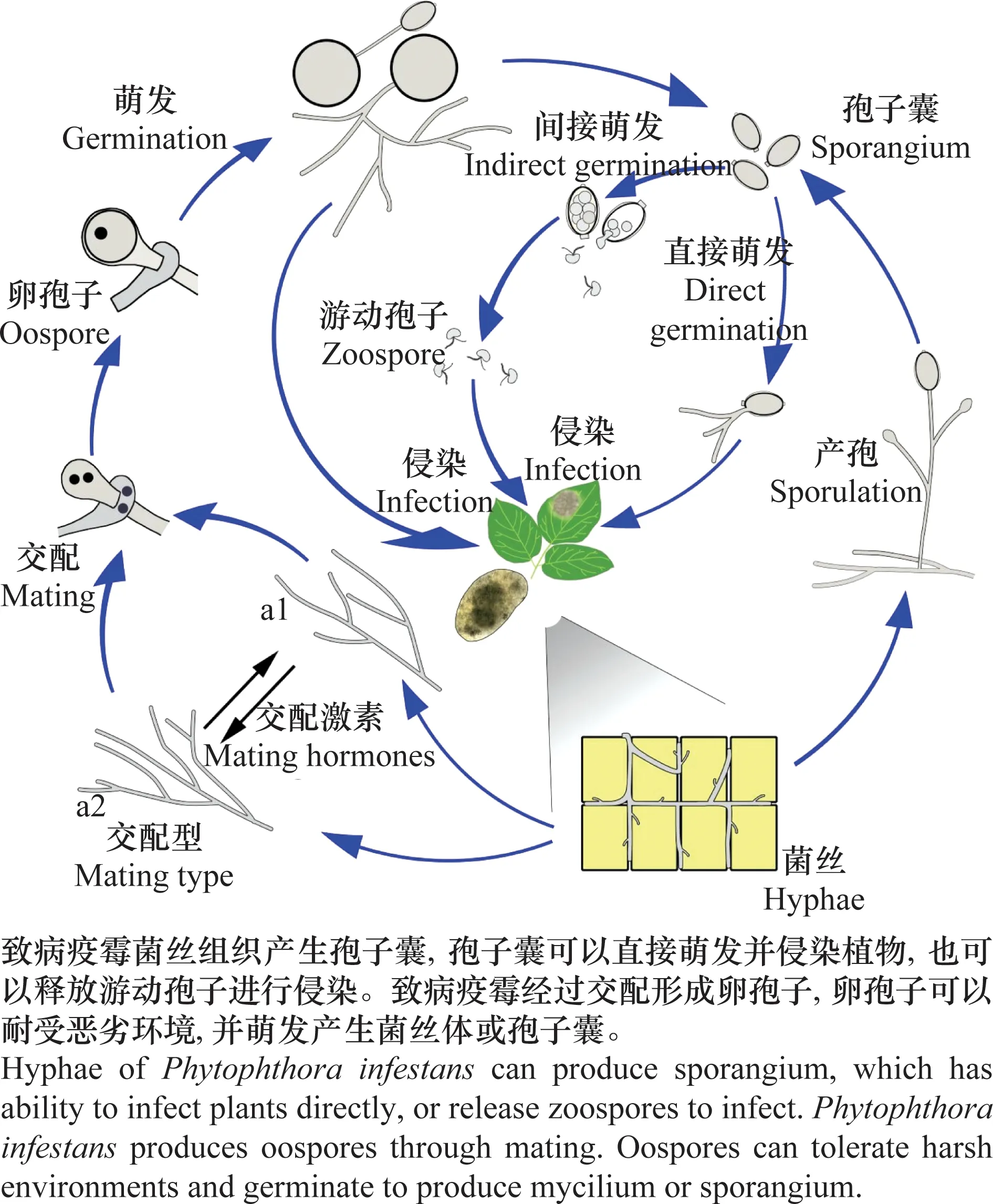
图1 致病疫霉的侵染循环Fig.1 Infection cycle of Phytophthora infestans
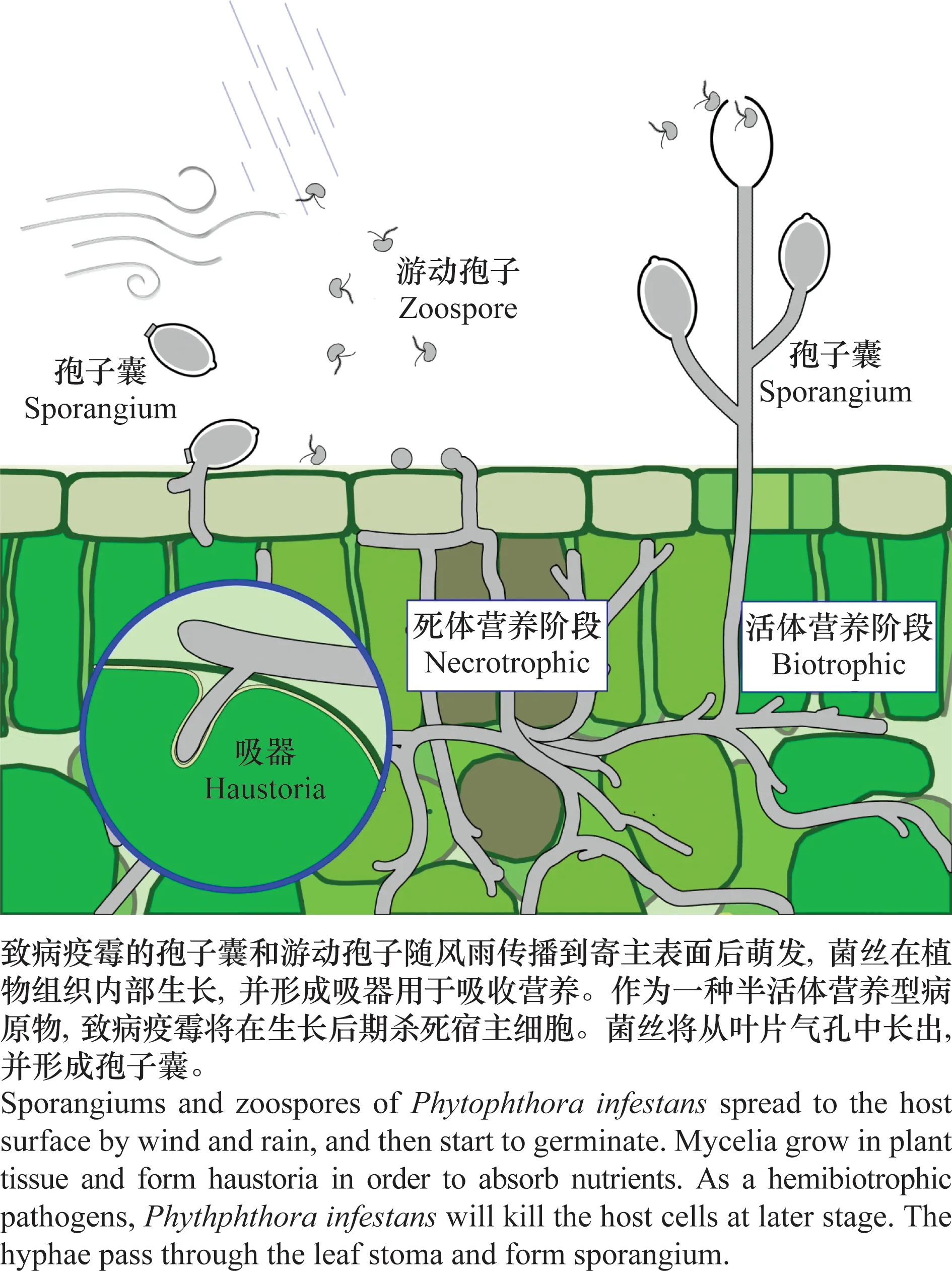
图2 致病疫霉的侵入、侵染和产孢过程Fig.2 Entry, infection and sporulation of Phytophthora infestans
致病疫霉通过有性生殖可以产生卵孢子(oospore),卵孢子能在土壤中长期存活,抵抗较恶劣的环境,作为下一个生长季节的初侵染来源,为晚疫病防治带来更多的压力(图1)[68-69]。有性生殖过程依赖于A1、A2两种交配型(mating type)菌株之间交配激素(mating hormones)的交流,而交配激素在疫霉属内是相对保守的,暗示种间杂交的可能性;此外,一些菌株可以同时分泌不同类型的交配激素[70-71]。A2交配型的菌株曾只在墨西哥被发现,直至1984年于瑞士被鉴定到[72];1996年,我国内蒙古自治区和山西省首次采集到A2交配型菌株[73]。近年来,我国致病疫霉种群结构处于快速变化中,各地菌株的种群结构差异极大;且在部分地区,能够自发产生卵孢子的自育型菌株已占据优势,这可能是由于自育型菌株具有更强的生活力和侵染能力[74-76]。未来,有必要持续关注各地致病疫霉种群结构变化,检测新型变异菌株的产生和流行,并对土壤中可能存在的卵孢子做预防处理。
致病疫霉的产孢过程涉及一系列信号通路的调节,G蛋白偶联受体(G protein-coupled receptor, GPCR)通路、丝裂原活化蛋白激酶(mitogen-activated protein kinase, MAPK)信号通路参与到疫霉不同类型孢子的产生过程中[77-80],近年研究还发现特定效应分子与致病疫霉产孢调节有关,例如4个RxLR效应分子基因在有性生殖过程中被诱导上调表达;而利用发夹结构敲低效应分子Avrblb2基因的表达量后,卵孢子的产生量随之显著下降[81]。一类在真核生物中非常重要的转录因子——MADS-box家族转录因子,也被发现是致病疫霉产孢所必需的,该研究为解析疫霉产孢调控机制和演化过程提出了新的思路[82-83]。进一步解析致病疫霉的生长调控机制,开展针对性的研究和管理,对于晚疫病防治具有积极意义。
2.2 致病疫霉的穿透与侵入
致病疫霉穿透寄主表面的过程涉及多种酶的作用,例如使用致病疫霉培养液中的半乳糖酶处理马铃薯细胞壁,可以分解高达23%的细胞壁组分[84],导致细胞壁强度下降;新的研究发现,一系列铜依赖的裂解多糖单加氧酶(lytic polysaccharide monooxygenase, LPMO)参与分解植物细胞壁中的果胶,沉默致病疫霉中的一个LPMO基因PiAA17C将导致几乎无法侵染,证明了化学机制在疫霉侵入过程中的重要性[85]。
近年来一系列研究逐步阐明了疫霉侵入植物表面的机械性机制。研究者注意到,致病疫霉菌丝侵入时总是倾斜的,而非真菌那样垂直侵入;随后他们使用透明的聚二甲基硅氧烷(psolydimethylsiloxane, PDMS)材质代替叶片以方便进一步观察。致病疫霉的游动孢子在PDMS表面表现出与在寄主植物上类似的生长特点,游动孢子休止后,萌发出芽管并黏附在基质表面,最终以约(49±10)°的倾角斜向突破基质表面。研究者观察到,芽管前部的基质材料出现延伸的裂缝,确认侵入过程中施加的力是斜向的;进一步的试验结果表明,这种斜向压力依赖于细胞骨架的重组。研究者把这种有别于真菌的倾斜侵入方式命名为“naifu”——这来源于日语中的“刀”一词,用以描述菌丝倾斜切入基质表面的过程[86]。后续研究发现,接触位置的菌丝中出现了放射状分布的肌动蛋白结构,其中心指向侵入凹痕,这种放射状结构来源于细胞骨架的重组,虽然其强度并不足以支撑侵入过程,但它充当了侵入过程中的自适应机械骨架,有助于平衡、调整侵入过程中的机械应力,从而维持侵入菌丝尖端的形状,避免侵入过程中尖端钝化,确保压力集中[87]。
致病疫霉侵入植物内部后,沿细胞间隙扩展并于菌丝上形成指状吸器(haustoria),深入附近植物细胞内部,有利于营养物质的吸收。近年的研究发现,吸器作为分泌的主要部位,通过常规与非常规的分泌手段,将一系列与侵染过程相关的蛋白质分泌到致病疫霉与植物的互作界面上,这些蛋白质包括PAMPs、胞壁降解酶、转化酶、效应分子等等,这一过程对于成功侵染是必需的[88-90]。吸器外侧包裹着一层植物细胞质膜衍生而来的膜,称为吸器外膜(extrahaustorial membrane,EHM),二者之间是被称为吸器外基质(extrahaustorial matrix)的狭窄空间。EHM上的膜蛋白与常规的细胞质膜上的蛋白存在很大差异,一部分质膜定位的蛋白质被排除在EHM之外,而另一些特殊蛋白质则被定向运输至EHM上,这一过程涉及植物囊泡运输系统的大规模重编程,并有可能同时受到病原菌的影响[91]。
疫霉侵染植物的根本目的在于吸收营养物质,在此过程中,吸器发挥着重要的作用。疫霉吸收营养的过程涉及到一系列营养转运体,包括负责转运糖、氨基酸、胆碱等小分子、硝酸盐和磷酸盐等离子的蛋白质。转录组分析显示,侵染过程中转运蛋白的表达表现出时间与空间上的特异性,例如一部分转运蛋白在侵染前期(叶片或薯块上)被强烈地诱导上调表达,并在4~6 d后快速下调;而另一个硝酸盐转运体基因PITG_13011在马铃薯叶片中发生大幅度上调表达,然而在块茎中接种时,这个转运蛋白的表达量则变化较小,表现出了空间上、器官上的表达特异性。沉默硝酸盐转运体基因将导致致病疫霉几乎无法侵染叶片,但对块茎的毒力仅仅受到很小的影响,而且在培养基上的生长模式不受影响,这证明硝酸盐转运体基因表达量变化的这种空间特征是其对特定侵染状况的适应。进一步的分析表明,环境中硝酸盐离子的浓度、光照条件都可能影响硝酸盐转运体基因的表达[92]。总体而言,全基因组范围内的转录情况分析显示转运体基因受到复杂的表达调控。
2.3 致病疫霉的效应蛋白功能研究进展
基因组、转录组等组学研究大大推动了致病疫霉效应分子的发掘和机制解析。第一个完整的致病疫霉基因组于2009年公布,该基因组大小约240 Mb,远超大豆疫霉Phytophthorasojae(95 Mb)和栎树猝死病菌Phytophthoraramorum(65 Mb)[93]。研究者从致病疫霉基因组中预测了17 797个蛋白,数量超过大豆疫霉的16 988个和栎树猝死病菌的14 451个。基因组学研究开启了效应分子研究的新阶段,研究者注意到致病疫霉的基因组是不均匀的,一部分区域密集编码保守基因,且重复序列含量很低;而在高密度区域之间存在基因密度较低、保守性差、重复序列丰富的稀疏区[93]。进一步研究表明,大量效应分子定位于基因组的稀疏区域,数量也远超大豆疫霉和栎树猝死病菌[93]。值得注意的是,这种基因组结构和基因密度分化引出了被称为双速基因组(two-speed genomes)的概念,该模型认为致病疫霉基因组中重复序列丰富、基因稀疏的区域通过更快的演化,形成庞大而多变的效应分子库,帮助病原菌侵染;此外,更快的演化速度有助于病原物逃避寄主植物的免疫反应[94]。目前,类似的基因组结构和演化情况已被证实存在于其他病原微生物中,例如大丽轮枝菌Verticilliumdahliae[95]和禾谷镰孢Fusariumgraminearum[96]。
2005年,一个重要的蛋白质基序——RxLR(精氨酸-任意氨基酸-亮氨酸-精氨酸)被发现,该基序广泛存在于包括致病疫霉在内的多种卵菌病原物的分泌蛋白中,通常位于蛋白质N端附近[97]。致病疫霉的完整基因组序列被公布后,研究人员根据保守基序和其他特征,在致病疫霉菌株T30-4全基因组范围内预测到563个RxLR效应分子[93]。RxLR基序广泛存在于卵菌病原物中,曾被认为介导效应分子向寄主植物细胞内的转运[98],然而近年研究发现,致病疫霉效应分子AVR3a的RxLR基序可能在分泌前被切除,且这不影响它转运至植物细胞中,说明我们对RxLR基序的功能、对效应分子转运机制的认知仍有不足[99]。另一类重要的效应分子被称为CRN效应分子,这一名称源自“Crinkling”和“Necrosis”,用于描述最早一批被鉴定的CRN效应分子,具有引起本氏烟Nicotianabenthamiana叶片萎缩、褶皱和坏死的能力[100]。CRN效应分子在N端50个氨基酸附近具有保守的LxLFLAK(亮氨酸-任意氨基酸-亮氨酸-苯丙氨酸-亮氨酸-丙氨酸-赖氨酸)基序,这一基序同样被认为介导效应分子向植物细胞内的转运[101]。致病疫霉T30-4基因组中已预测到196个CRN效应分子[93],这些成果开启了致病疫霉的效应分子组学研究,在此后致病机制和互作研究中发挥了关键作用。
效应分子的分泌过程主要发生于吸器,且不同效应分子涉及不同的分泌机制,例如利用蛋白质转运抑制剂布雷非德菌素A(Brefeldin A, BFA)处理菌丝,抑制从内质网到高尔基体的运输过程,将会导致胞间效应分子PiEPIC1的分泌被完全阻断,然而胞内效应分子Pi04314的分泌不受影响,表明在致病疫霉中,胞内效应分子可能遵循非传统的分泌方式[88],这一点与先前在稻瘟病菌Pyriculariaoryzae上的相关研究结论一致[102]。进一步阐明效应分子的分泌机制,对于解析致病疫霉致病机理、筛选用于监测和防治的关键位点,将具有重要意义。
来源于致病疫霉88069菌株的CRN效应分子CRN8在侵染期间被转运至宿主细胞核中,在本氏烟叶片上表达CRN8,可以导致叶片在5 d后坏死;然而瞬时表达CRN8 1 d后接种致病疫霉,则发现CRN8对致病疫霉的侵染有促进作用;并且CRN8促进侵染和引发坏死的能力都依赖于它的激酶结构域[103]。此外,对辣椒疫霉Phytophthoracapsici的研究显示,相当一部分CRN效应分子并不会引发坏死,且促进了病原物对本氏烟的侵染[104];对大豆疫霉上的研究同样证实了CRN效应分子使病原菌毒力增强,且发现一个CRN效应分子CRN78靶向植物细胞质膜上的水通道蛋白[105]。目前,针对致病疫霉中CRN效应分子功能的研究相对较少,作用靶标和机制有待进一步发掘。
效应分子的后续作用机制多样而复杂,最显而易见的证据在于不同效应分子进入植物细胞后亚细胞定位存在很大差异,例如对52个致病疫霉RxLR效应分子的检验显示,其中41%存在于细胞核与细胞质,25%仅定位于细胞核中,18%靶向细胞质膜[106]。利用coIP/MS分析RxLR效应分子靶向的互作蛋白,发现这些效应分子可能涉及复杂的功能,包括蛋白质翻译、代谢调控、光合作用、囊泡运输等[107]。在这里,我们汇总了近年研究比较清楚的致病疫霉效应分子及它们的作用靶标和功能(表1)[61-62,64,66,108-127]。

表1 主要已知致病疫霉效应分子及其功能Table 1 Main effectors of Phytophthora infestans and their functions
许多效应分子作用于寄主免疫相关联的靶标和信号通路。MAPK信号通路作为调节真核细胞生理过程的重要信号通路,受到Pi13628、Pi17316、Pi21422、Pi22926等一系列效应分子的影响[66,119,122,125],有趣的是Pi21422和Pi22926分别靶向StMAP3Kε和StMAP3Kβ2,而这二者在MAPK信号通路中的作用是平行的,致病疫霉通过两个效应分子分别靶向这两个位点。蛋白质的合成过程也受到大量效应分子的直接或间接调控,其中Pi23226通过直接抑制寄主核糖体的生物合成影响全局水平的蛋白质翻译[126];蛋白质合成过程中,mRNA前体发生选择性剪接(alternative splicing)以产生多样的转录本,剪接因子(splicing factors)参与这一过程,而一些致病疫霉效应分子靶向剪接因子,影响植物细胞大量选择性剪接事件的发生[61];另一个效应分子Pi03192特异性定位于寄主的内质网膜,其靶标为NAC转录因子,这些转录因子在特定刺激下会从内质网膜上释放,并重定位至细胞核内,调控后续转录;Pi03192通过阻止内质网上的NAC转录因子释放,影响它们后续的重定位,来干扰相关信号通路、影响特定蛋白质合成[110-111]。可见,病原菌的“攻击”是多层次、大范围的,从转录调控、选择性剪接、翻译等多个水平上影响蛋白质的合成。
利用新分析方法和试验手段,从不同的思路发掘新型效应分子,已成为病原物研究的重要方向。Huang 等设计了一个选择性剪接报告系统,该系统基于一个番茄的类受体蛋白激酶(receptor-like protein kinase, RLPK),后者经历选择性剪接事件,且在免疫过程中发挥重要功能。研究者将RLPK基因序列与荧光素酶报告基因(luciferase, LUC)串联后,使用35S启动子在本氏烟中表达,在通常情况下,该重组序列产生RLPK-LUC全长转录本,然而一些效应分子将会导致RLPK在剪接中发生内含子保留,在新转录本中保留一个提前终止密码子,无法正确表达LUC,经荧光素底物处理后,表现为荧光强度下降[61]。研究者利用该报告系统筛选了9个影响选择性剪接的RxLR效应分子,大大推动了相关研究,这一思路值得我们学习与借鉴,可以根据可能的效应分子功能,选取新的靶标蛋白,设计其他报告系统用于筛选影响特定功能的效应分子。2019年,Thilliez等将基因富集测序技术应用于致病疫霉分泌蛋白分析,提出了病原富集测序(pathogen enrichment sequencing, PenSeq),证明该技术在分析效应分子的多态性和变异情况、检测特定基因的等位基因或同源基因、预测新的效应分子等方面大有可为[128];2020年,Lin等利用cDNA病原富集测序,结合长读长测序(long-read sequencing)技术,分析不同致病疫霉菌株之间的效应分子表达差异,并鉴定了被少花龙葵Solanumamericanum抗病基因Rpi-amr1识别的致病疫霉效应分子Avramr1(Pi07569)[129]。可以预见,新技术和新分析方法的应用将大大加速致病疫霉的研究,鉴定更多的关键位点,加深我们对疫霉-植物互作机制的理解。
值得注意的是,许多效应分子的毒性功能仍有待解析,这方面体现在信号通路的上下游仍不完全清楚,一方面则在于一些基于本氏烟的研究成果缺乏在野生寄主(马铃薯和番茄)上的验证。深入发掘更多效应分子的毒力机制,还原田间侵染过程中的互作情况,将是后续研究的重要方向。
2.4 新型毒力因子的发掘工作
一些新兴的病原物-植物互作机制引发了学界关注。跨界小RNA能通过靶向其他物种的特定mRNA,引起基因沉默,其在病原物-植物互作过程中也发挥了一定的作用。一个来自致病疫霉的小RNA——miR8788,已被证明诱导马铃薯水解酶基因StABH1沉默,该基因对于马铃薯抗晚疫病有重要作用[130]。类似的机制在其他病原物中早已被发现[131-132]。相对应的,植物也利用小RNA沉默病原菌中与毒力相关的基因,从而对抗病害[133]。Luo等从马铃薯全基因组范围内预测了53个可能靶向致病疫霉的miRNA,其预测靶标包括转录因子、跨膜蛋白、激酶等,其中一部分,例如miR394的瞬时表达可以抑制致病疫霉的侵染,然而一部分预测的miRNA例如novel133、novel166和miR396过表达后对致病疫霉的侵染表现为促进作用,它们在互作过程中所处的地位还有待更多研究[134]。有趣的是,一部分miRNA同时靶向马铃薯和致病疫霉的基因[134]。2022年,Chi 等开发了一套算法用于预测可能在植物和病原物互作过程中发挥了跨界调节作用的小RNA,目前尚未被应用于致病疫霉相关研究[135],我们期待未来的进一步研究。
磷脂也可能作为毒力因子参与疫霉的侵染过程。大豆疫霉的效应分子Avh5 向寄主细胞内的转运依赖于其与磷脂酰肌醇3-磷酸的结合[136],而Lu等的研究表明,大豆疫霉在侵染过程中会分泌额外的磷脂酰肌醇3-磷酸,这些磷脂对于效应分子的转运和疫霉侵染有促进作用;而植物的磷脂酰肌醇3-磷酸结合蛋白可以增强抗病性[137]。这一互作机制可以应用于抗性育种,通过将磷脂酰肌醇3-磷酸结合蛋白与外源抗真菌蛋白融合,可以培育出具有持久广谱抗病性的转基因植物,具有广阔的应用前景[138]。
近年来,多组学分析大大推进了毒力因子的发掘工作。2010年,Raffaele等根据基因的N端信号肽、跨膜序列等特征,预测致病疫霉全基因组范围内的分泌蛋白,形成预测的分泌蛋白质组,并根据保守基序和基因本体(gene ontology, GO)分析的功能预测结果,对分泌组蛋白进行了归类和注释;随后,根据基因在致病疫霉基因组上的位置(位于基因密集区域或稀疏区域),结合不同侵染阶段的转录组数据以及不同疫霉的基因组比较结果,在致病疫霉全基因组范围内预测了一系列候选的毒力基因,包括编码一系列外泌酶、富含半胱氨酸的小分子分泌蛋白、含重复序列的分泌蛋白等的基因[139]。Seidl等结合基因结构与转录组数据,对致病疫霉的启动子和转录因子进行了系统分析,预测了4个在致病疫霉侵染过程中参与调控基因上调表达的特殊DNA基序[140],有助于后续毒力基因的预测和筛选。
Chen 等鉴定到大豆疫霉和致病疫霉中的一类重要的表观修饰酶——6mA甲基转移酶,随后发现DNA的6mA甲基化修饰广泛存在于这两种疫霉的基因组中,致病疫霉和大豆疫霉分别有1 805和1 343个基因存在6mA标记;6mA修饰与相应基因的表达量表现为负相关;敲除大豆疫霉中的6mA甲基转移酶基因引起超过3 000个基因的表达量发生变化,涉及代谢和侵染过程中的重要基因,并且导致大豆疫霉的毒力下降,暗示疫霉毒力因子受到表观遗传调控[141]。随后,研究者在疫霉中鉴定到更多的表观修饰,并绘制了疫霉组蛋白H3第27位赖氨酸三甲基化(H3K27me3)和第36位赖氨酸三甲基化(H3K36me3)在基因组范围内的分布图,发现表观遗传修饰对于调控基因组范围内的基因表达,调节转座子活动和维持基因组的稳定性有重要意义[142-143]。Miao等发现,6mA修饰对于灰霉病菌Botrytiscinerea的致病能力同样具有重要意义[144],也印证了从表观基因组学层面开展致病疫霉毒力机制研究的重要性。
2018年,Rodenburg等将致病疫霉全基因组内的预测蛋白质与已有数据库比对,并根据保守的催化结构域等特征进行功能预测和模型训练,预测可能发生的代谢反应。共1 408个基因被预测参与1 569种不同的生化反应,其中涉及1 663种代谢物;随后,通过计算每个生化反应的通量,结合涉及的酶的亚细胞定位预测数据,经过纠错、整合得到了第一个致病疫霉全基因组水平的代谢模型(genome-scale metabolic models,GEM),这一模型包含2 394个生化反应,可以对致病疫霉不同阶段的代谢情况进行定性预测,为后续研究不同阶段的代谢调控状况提供了便利[145]。Botero等也建立了类似的代谢模型,并从侵染的角度进行分析,根据不同时期的转录组数据,建立了寄主组(在培养基上不活跃,在接种早期活跃的代谢反应)、活体营养组(在侵染后2~3 d活跃、在4 d后下调的代谢反应)、死体营养组(在侵染后2~3 d不活跃、在4~5 d上调的代谢反应)以及过渡组(活体营养阶段向死体营养阶段过渡过程中发生显著变化的代谢反应)4个特异性代谢组,并以此分析每个阶段的代谢模式和特征反应,鉴定了一系列在侵染过程中受到关键调控的基因[146],进一步的分子互作和机制研究有待进一步展开。
新型的致病疫霉毒力因子大大丰富了我们的认知,而在此过程中,多组学方法的广泛应用有助于发掘以前未知的作用机制。我们期待利用新的试验与分析手段,鉴定到更多新型致病因子,并对后续信号通路和分子机制进行更深层次的研究,完善我们对互作体系的认知,最终指导晚疫病防治。
3 寄主识别致病疫霉的机理研究
3.1 致病疫霉的PAMPs及寄主PTI反应研究进展
植物的模式识别受体(pattern recognition receptors,PRRs)识别病原相关分子模式(pathogen-associated molecular patterns,PAMPs)后会引发一系列生理反应。包括但不限于活性氧(reactive oxygen species,ROS)迸发[147],细胞膜电位变化[148],GPCR信号传递[149],以及后续的转录调控和免疫激活,这些反应共同构成了植物的病原相关分子模式触发免疫(pathogen-associated molecular patterns-triggered immunity,PTI)。值得注意的是,前期在研究拟南芥ArabidopsisthalianaPTI反应时发现,一个类受体激酶SERK3/BAK1是拟南芥PTI反应的中枢核心,本氏烟中的同源蛋白NbSerk3/Bak1对于本氏烟响应一系列不同来源的PAMPs触发的免疫反应也是必需的[150]。
INF1是一个经典的PAMP激发蛋白,在致病疫霉的菌丝阶段表达,但在侵染阶段,尤其是侵染早期的活体营养阶段下调[151]。早期研究证明,本氏烟通过识别INF1介导对致病疫霉的抗病性[152],且这种识别同样依赖于NbSerk3/BAK1[153],与拟南芥中的PTI反应类似。马铃薯的激发素类受体蛋白ELR也参与了对INF1的识别和对致病疫霉的免疫反应。ELR属于类受体蛋白(receptor-like protein,RLP),可以与SERK3/BAK1发生分子间互作,且这种互作会随INF1处理而增强[154]。
另一个著名的疫霉PAMP分子是XEG1,该蛋白质最早在大豆疫霉中被发现。XEG1属于糖苷水解酶12(glycoside hydrolase family 12,GH12)家族,具有木葡聚糖酶和β-葡聚糖酶活性,且靶向质外体,这提示它参与对寄主植物细胞壁的降解;沉默XEG1导致大豆疫霉的毒力下降,证明了XEG1对侵染的促进作用,但与此同时,过表达XEG1的大豆疫霉侵染能力也受到了损害,且植物组织中ROS迸发和胼胝质积累远超对照菌株,说明这种侵染能力的下降可能源自植物免疫反应的加强。XEG1触发的PTI同样依赖于SERK3/BAK1。此外,致病疫霉中预测到6个XEG1同源基因,但其中只有一个能在本氏烟上引起细胞死亡[155]。有趣的是,大豆分泌的抑制因子GmGIP1可以特异性结合大豆疫霉XEG1,从而抑制后者的糖苷酶活性;然而大豆疫霉基因组中存在一个截短的XEG1旁系同源蛋白XLP1,该蛋白不具有糖苷酶活性,但与GmGIP1结合能力远超XEG1,这使得XLP1可以通过消耗植物分泌的GmGIP1来保护XEG1不被抑制,从而维持大豆疫霉的毒力水平。在这个模型中,XLP1作为诱饵,捕获植物分泌的抑制蛋白并将其中和[156]。在寄生疫霉中,研究者发现PsXEG1的同源蛋白PpXEG1,以及另一个全长GH-12家族糖苷水解酶PpXLP1,以相似的机制对抗本氏烟分泌的抑制因子NbGIP2,尽管NbGIP2与GmGIP1并非同源,这说明这种互作模式可能是广泛存在的,并在不同物种中表现出趋同演化倾向[156]。
烟草中的一个富亮氨酸重复结构的类受体蛋白(leucine-rich repeat-receptor like protein, LRR-RLP)可以识别大豆疫霉的XEG1,并因而被命名为RXEG1。RXEG1介导的抗病性依赖于BAK1和SOBIR1两个类受体激酶(receptor-like protein kinase, RLK)的参与,证明RLK在植物PTI反应中占据特殊地位[157-158]。NbRXEG1甚至可以识别小麦上的禾谷镰孢F.graminearum编码的GH12糖苷水解酶,尽管本氏烟并非禾谷镰孢的天然寄主,显示这类PAMPs在远缘植物病原物中分布广泛,且结构和功能上具有一定的保守性[159]。
致病疫霉基因组编码65个富含半胱氨酸的小蛋白(small cysteine-rich, SCR),其中一个SCR蛋白,PITG_14439(被命名为PC2),已被证明可以作为PAMP被植物识别,引发后续的ROS迸发和细胞死亡。植物对PC2的识别依赖于质外体丝氨酸蛋白酶(例如番茄丝氨酸蛋白酶P69B)对其进行切割,而一个致病疫霉分泌的蛋白酶抑制剂EPI1可以抑制这种切割,从而抑制相关免疫反应的发生。这一研究提示,植物对PAMPs的识别存在复杂的机制,包括识别前对PAMPs的“预处理”步骤,而病原体可能针对这些步骤进行干扰和阻碍[160]。
Pep-13是一个在卵菌中高度保守的基序,最早从大豆疫霉的一个具有谷氨酰胺转氨酶(transglutaminases, TGase)活性的糖蛋白GP42中被鉴定到,且可以作为PAMP被马铃薯识别,触发PTI反应[161]。Pep-13引发的PTI反应不依赖SERK3/BAK1[162];参与识别Pep-13的PRR基因定位于马铃薯3号染色体顶端附近,该区域中曾被注释到5个候选LRR-RLKs,然而进一步的试验表明这5个RLKs都不能单独介导针对Pep-13的PTI反应[163],这可能是因为相关基因的开放读码框尚未被注释到,也可能由于针对Pep-13的PTI反应并非由单一RLK介导。
需要注意的是,以上几种PAMPs均属于蛋白质或肽类,而病原菌的其他分子也可以作为PAMPs被植物识别。一些脂肪酸,例如来自致病疫霉的二十碳五烯酸和花生四烯酸可以激发马铃薯的免疫反应,引起倍半萜等次生代谢产物的积累[164],引发马铃薯对致病疫霉的抗性增强[165]。此外,真菌细胞壁中的多糖组分,例如几丁质(chitin),也可以作为PAMPs触发植物的PTI反应[166-167],但目前尚无证据证明卵菌细胞壁对植物PTI的激发作用。非蛋白质类PAMPs分子在卵菌-植物互作过程中所处的地位,仍有待进一步研究。
3.2 抗病基因介导的ETI
抗病基因(resistance gene,Rgene)表达后识别病原菌的效应分子,引发后续包括过敏反应(hypersensitive response,HR)在内的一系列免疫应答,称为效应分子触发免疫(effector-triggered immunity,ETI);在此过程中,被识别的效应分子也被称为无毒基因(avirulence gene,Avr)。在早期的晚疫病抗病育种研究中,最初的抗病基因主要来自于墨西哥六倍体野生种马铃薯Solanumdemissum,其所含的11个(R1-R11)抗晚疫病基因,均为小种专化型的主效抗病基因[168-169]。但随着致病疫霉各生理小种的不断变异,早期的抗病基因已全部失去抗病功能[170]。随着现代分子生物学与高通量测序技术的快速发展,利用抗病基因图位克隆结合比较基因组学,抗性基因富集测序(resistance gene enrichment sequencing, RenSeq)以及病原菌效应子组学等方法,抗病育种学家不断从各类野生马铃薯资源中克隆到全新的抗病基因,最著名的广谱抗性基因包括Rpi-vnt1.1、Rpi-blb1、R8、Rpi-amr1等[171-174],但能够克服这些抗病基因的致病疫霉生理小种也随之涌现[175]。此前,Paluchowska等[176]已对茄属Solanumspp.植物中的抗晚疫病基因进行了系统的整理,我们不再重复罗列。在这里,我们把目光移向近年来新发现的抗病基因识别与作用机制,关注这一领域的理论进展。
NLR(nucleotide-binding domain and leucine-rich repeat-containing)是最重要的抗病基因家族,一般认为其LRR结构域是决定识别特异性的关键,例如茄属植物中抗病基因Rpi-chc1的一系列等位基因在LRR结构域上存在分化,而这种分化使得它们能够识别致病疫霉PexRD12/31超家族中的一系列效应分子[177];又如马铃薯的两个抗病基因Gpa2和Rx1分别可以识别马铃薯白线虫Globoderapallida和马铃薯X病毒(potato virus X),人为重组它们的LRR结构域可以使它们的识别功能发生交换,而二者的其他结构域和内源启动子的交换均不会影响识别功能[178]。然而,NLR对效应分子的识别功能并不一定依赖于LRR结构域,一个极端的案例是拟南芥中的RBA1,该抗病基因缺失了常规NLR中保守的NBS和LRR结构域,仅有一个Toll/白介素1样受体(Toll/interleukin-1 receptor, TIR)结构域,然而RBA1可以完成对丁香假单胞菌Pseudomonassyringae效应分子HopBA1的识别[179]。
值得注意的是,以上识别方式依赖于抗病蛋白和效应分子的直接结合,但近年的一些研究显示,某些效应分子触发的ETI反应涉及间接识别。研究者们提出了一系列模型用于解释这些间接识别过程。“守卫(Guard)模式”最早被用于描述番茄对丁香假单胞菌效应分子AvrPto的识别,在这个模型中,AvrPto靶向番茄防卫蛋白PTO,而PTO与番茄NLR蛋白PRF存在组成型的分子间互作,AvrPto对PTO的识别“惊扰了”PTO,导致PTO与PRF解离,激活下游的ETI信号通路[180-181]。在这个模型中,抗病蛋白并未直接识别效应分子,而是“监控”效应分子靶向的关键靶标,通过这些靶标的变化发现前来入侵的效应分子。这一模型的基本逻辑是,抗病蛋白所监控的靶标位点在植物免疫反应中是存在特定功能的,并因此才会成为病原菌效应分子靶向的目标,然而这种功能上的两面性将导致这类靶标位点受到相反的选择压力,并很可能表现出分裂选择的演化模式:要么避免被效应分子靶向,逃避病原菌的攻击;要么增强与效应分子的结合能力,确保后续ETI反应足够敏感。后续的研究发现,病原菌效应分子可能攻击不止一个靶标,而并非所有的靶标都有助于增强毒力,例如拟南芥RIN4受到丁香假单胞菌效应分子AvrRpt2的直接靶向,并且参与后续ETI反应,然而RIN4的T-DNA插入失活突变体上AvrRpt2的毒力功能非但没有减弱,反而有所增强,这与“守卫模式”的基本逻辑相违背[182]。基于这些推理和试验结论,研究者于2008年推演了“守卫模式”向“诱饵(decoy)模式”的转变,在诱饵模式中,病原物效应分子的靶标并非唯一的,而真正参与后续ETI反应的靶标作为诱饵存在,攻击诱饵对植物免疫的损害有限或根本没有损害,却可以激活后续抗病基因的识别[183]。在此基础之上,Cesari等于2014年提出了新的“整合诱饵(integrated decoy)假说”,他们注意到一些抗病蛋白携带有额外的、非保守的结构域,且这些结构域往往涉及和效应分子的互作,例如抗病蛋白RRS1带有WRKY结构域,可以与效应分子AvrRps4 结合;而另一个抗病蛋白Pi5-2在C末端具有效应分子AvrRpt2的识别位点,基于这些证据,研究者推测,一些“诱饵模式”中的诱饵关键识别位点已被整合到抗病蛋白上,作为“整合诱饵”,从而允许抗病基因直接识别效应分子[184-185]。这一猜测提示我们,可以将效应分子的作用靶标与已知的NLR蛋白整合,来人工“定制”抗病基因、识别其他效应分子。例如用能够识别荧光蛋白的单结构域抗体片段(single-domain antibody fragment)替换Pik-1的整合结构域,使得重组后的Pik可以识别荧光蛋白、引发坏死,这无疑为抗病资源的发掘应用提供了全新的思路[186]。
一组被称为NRC(NLR required for cell death)的NLR抗病基因早就引起了研究者的关注。NRC1最早于番茄对叶霉Fulviafulva的免疫反应中被发现,它参与了番茄抗病基因Cf-4对叶霉无毒基因Avr4的识别,并且参与到Avr9、AvrPto(来自丁香假单胞菌P.syringae)、tvEIx(来自绿色木霉Trichodermaviride)等一系列效应分子引发的HR反应过程中[187-188],这种“一对多”的功能特点不同于传统的基因对基因学说。更多的研究证明,许多(但不是全部)互作案例中存在另一个不参与识别,但对坏死功能至关重要的NLR蛋白存在;这种情况下,参与识别无毒蛋白的NLR被称为“Sensor NLR”,而不参与识别、辅助信号传导的NLR被称为“Helper NLR”[188-189]。随后,NRC2、NRC3和NRC4被鉴定到,且研究发现这些Helper NLR在信号传递中的功能有时是可以互相替代的,例如马铃薯抗病基因Rx介导对马铃薯X病毒(PVX)的效应分子CP的抗性,而这种抗性可以依赖NRC2、NRC3或NRC4中的任意一者实现,只有当这3个Helper NLR全部被沉默后,才会表现出抗性受损。这种“冗余性”机制确保了植物ETI反应不会因少数Helper NLR出现异常而遭受破坏,大大提高了植物免疫的稳定性[190-191]。当然,并不是所有NLR都需要成对发挥作用,一个有趣的NLR,拟南芥中的ZAR1揭示了全新的机制。ZAR1能够识别来自多种病原物的不同效应分子,其识别过程涉及一系列RLKs参与,例如在识别野油菜黄单胞杆菌Xanthomonascampestris效应分子AvrAC的过程中,AvrAC通过尿苷化PTI过程中的一个RLK——BIK1发挥毒性功能,但在此过程中被并不参与PTI的诱饵PBL2捕获;尿苷化的PBL2与ZAR1和另一个假激酶RKS1形成复合体,并经寡聚化形成一个五聚体,被称为抗病小体(resistosome)[192-193]。该抗病小体呈漏斗形,可以嵌入细胞膜,成为钙离子通道,带来快速的钙离子内流,并最终触发免疫反应和细胞坏死[194]。在这个实例中,ZAR1同时起到了识别和信号传递的功能,且通过寡聚化、形成离子通道快速启动细胞坏死。
NLR的寡聚化和抗病小体的形成已被发现在多个抗病过程中发挥重要作用,例如在少花龙葵抗病基因Rpi-amr3对致病疫霉效应分子AVRamr3的识别过程中,Rpi-amr3诱导辅助NLR蛋白NRC2与NRC4寡聚化形成抗病小体,介导下游的免疫反应;而在另一个抗病基因Rpi-amr1对无毒基因AVRamr1的识别过程中,仅有NRC2被诱导形成抗病小体[195],这一研究证明抗病小体在晚疫病抗病过程中发挥重要而复杂的作用。
另一个值得注意的问题是,尽管传统的zig-zag模型将PTI与ETI描述为泾渭分明的两个阶段,然而实际的植物抗病过程中,二者的关系是复杂的。辛秀芳研究员团队发现,阻断PTI会严重损害ETI的强度,因为PRR相关的信号传递是ETI所必需的,且ETI过程中的ROS产生依赖于来自RLK的信号,这证明PTI是ETI的基础[196];而与此同时,ETI与PTI会相互增强,这涉及一系列关键基因的转录增强和积累[197],提示我们不能将两种免疫过程简单拆分讨论,我们期待未来致病疫霉与马铃薯的互作研究将带来更多基础理论更新,进一步丰富我们对植物病理学的了解。
随着长读长三代测序技术的不断进步与抗病基因高精度注释工具的开发完成[198],未来的抗病基因挖掘必将立足于针对高抗性马铃薯品系基因组数据的完善分析与对抗病基因进化历程的深入理解。此外,我国并非马铃薯的起源地,晚疫病抗性资源相对匮乏,广泛种植的马铃薯品种背景狭窄,抗病资源库匮乏,即使有少数高抗晚疫病的资源,但对其抗病遗传背景和基因组信息知之甚少,极大限制了对抗性资源的有效利用与开发,所以,广泛地搜集世界各地高抗晚疫病马铃薯品种,进行大规模的高通量测序,深入开展抗病基因组学分析是解决这一问题的关键。全基因组关联研究(genome-wide association studies,GWAS)允许研究者在全基因组范围内筛选与特定表型相关的遗传位点,Juyo Rojas 等利用该技术筛选了16个马铃薯中与晚疫病抗性有关的位点[199];Jupe等利用富集测序技术系统性地寻找茄属植物中的NB-LRR基因,在马铃薯和番茄中鉴定到了大量此前未被注释的NLR基因[200-201]。新技术的应用,大大降低了抗病基因的发掘与鉴定难度,极大地推动了相关领域的研究进展。
4 致病疫霉对寄主ETI的应对
病原菌面对ETI反应并不会“束手就擒”,而是采取多种手段应对。最直接的应对方式是,无毒基因可能发生序列突变以逃避抗病基因的识别,例如致病疫霉效应分子Avr1的一个同源基因A-L逃避了抗病基因R1的识别,不会引发HR[202]。事实上,Avr1具有丰富的多态性,例如Shen等从来自全国的111个致病疫霉菌株中鉴定到了49个氨基酸序列存在差异的Avr1同源蛋白,且存在一个高频突变T512G,该突变导致Avr1序列提前终止,末端的38个氨基酸被截断,这种被截断的Avr1蛋白并不会引发HR,从而逃避R1的识别。然而,这种应对是有代价的,截断T区后的Avr1同时也将失去靶向Sec-5、阻断PR-1分泌的毒性功能,尽管如此,对于病原菌而言,这种突变总体上是值得的[121,203]。一系列其他致病疫霉效应分子也存在类似的策略,包括Avr2[204]、Avr3a[205]、Avr4[63]等,均存在多样的同源基因。致病疫霉特殊的基因组结构,以及“双速基因组”假说,有助于解释这种广泛的效应分子多态性的来源[94]。
另一种常用的策略是调控效应分子的表达。Pais等的研究证明致病疫霉不同菌株间存在很高的基因表达多态性,而部分菌株中,一个重要的无毒基因Avrvnt1(Pi16294)受到沉默,使得这些菌株在携带抗病基因Rpi-vnt1的马铃薯上恢复了毒力[206];在其他病原菌的研究中已经发现病原菌通过小RNA沉默、表观遗传调控等方式抑制效应分子表达,从而逃避抗病基因的识别[207]。事实上,效应分子的表达受到复杂的调控,Ochola等的研究证明,效应分子的表达调控,尤其是其在侵染不同阶段的差异表达,对于植物抗病基因识别和免疫反应具有极大的影响,可能是病原菌对抗植物ETI反应的重要方式之一[208]。
通常认为,效应分子靶向植物的基础免疫过程,然而在其他植物病原物上的研究证明,效应分子同样可以靶向ETI,揭示了植物-病原物之间“军备竞赛”的进一步发展。Nakano等发现4个来自青枯病菌Ralstoniasolanacearum的效应分子RipI,RipAC,RipAP和RipAU可以抑制另一个效应分子RipAA在本氏烟上引发的HR反应。随后,他们重点关注RipAC,并证明RipAC通过靶向烟草NbSGT1,抑制了ETI信号通路,而后者正是RipAA引发坏死所必需的。最后,研究者证明了RipAC有助于青枯病菌在抗性本氏烟上生长[209]。在该研究中,效应分子靶向植物ETI途径而非通常的PTI,类似的机制目前尚未在致病疫霉与寄主的互作过程中被证实,有待学界进一步关注。植物与病原物的“军备竞赛”对抗病基因的推广利用提出了更高要求,发掘具有持久抗病能力的抗病基因、监测田间菌株的毒力变化,是植物病理学研究的长远目标。
5 总结与展望
马铃薯晚疫病严重威胁马铃薯生产,挑战我国马铃薯主粮化战略和粮食安全。当前阶段,我国马铃薯发病面积大,危害严重,防控晚疫病的需求紧迫[13-14]。化学防治仍然是我国晚疫病防治的重要手段,然而大量施用化学农药与我国农药减量化行动方案相悖,且近年来,田间抗药性菌株频发,极大地挑战了化学防治的效果[210]。开发、推广新的晚疫病防治手段,是我国粮食生产的重要需求。
近年来,植物病理学互作机制和基础理论研究正在指导实际生产。许多抗病基因已经被应用于马铃薯抗晚疫病育种[176],且从非寄主中可能筛选到难以被克服的高效抗晚疫病基因[211]。作物抗病性往往以牺牲产量为代价,而在水稻中的转录因子IPA1有助于植物平衡产量和抗性水平,在正常状态下调控穗发育相关基因的表达、增加产量,而面临稻瘟病侵染时发生磷酸化,激活抗病相关基因的表达,及时提高抗病性[212],这一机制对于抗病品种的产量保障有重要意义,亟须在抗晚疫病育种中发掘类似的资源。此外,理论研究揭示了新的抗病思路,例如施用壳聚糖可以激发马铃薯的免疫反应,并提高其对致病疫霉的抗病性[213];喷洒以黏土作为载体,以致病疫霉各生理阶段的重要基因为靶标的RNA,能极大地抑制晚疫病发生[214]。一些生防菌对于晚疫病防治具有一定效果[215-216],值得进一步开发与推广。
晚疫病防治方法的突破,很大程度上依赖于植物病理学理论革新,病原物与寄主植物的互作机制一直是植物病理学研究的重点。近年来,对于致病疫霉的相关研究取得了不菲的成果,从穿透、定殖,到病原菌与植物间的军备竞赛,均有一系列新机制被发现,PTI与ETI的相互联系、效应分子对ETI的抑制作用、miRNA跨界调控等一系列发现大大丰富了植物病理学基础理论,在传统zig-zag模型的基础上增添了新的内容,这些新理论也为未来致病疫霉-马铃薯互作机制的进一步研究提供了全新的思路。同时,新的互作理论提醒我们,致病疫霉与植物的互作机制复杂程度远超我们的想象,新抗性资源的发掘、高效抗病基因的筛选、病原菌克服抗性情况监测是长期的需求,这对晚疫病快速检测、基因分型和分子标记等方面的研究提出了更高要求。
高效遗传转化体系的建立和优化对于后续研究有重要意义。在模式植物拟南芥的研究中,覆盖全基因组的T-DNA插入突变体库被广泛应用于基因功能研究,大大推动了基础理论的发展[217-218]。1991年,Judelson等首次开发了致病疫霉遗传转化体系,该体系利用聚乙二醇和氯化钙处理致病疫霉原生质体,获得了转化子[219]。2003年,基因枪法被应用于致病疫霉转化,该方法利用携带目的基因的金微粒轰击目标组织,从而获得转化子,不再需要制备原生质体;选用该方法时,最好的组织材料为孢子囊[220]。同时,Vijn等开发了农杆菌介导的致病疫霉转化体系,在乙酰丁香酮(acetosyringone)处理下,携带目的基因的农杆菌与致病疫霉的游动孢子共培养,可以得到致病疫霉转化子[221]。最近,Wang 等改良了这一流程,通过在农杆菌中引入来自拟南芥的AtVIP1基因,大幅度增加了转化效率[83]。高效遗传转化体系的建立,为基因编辑株系的培育和基因功能研究奠定了基础。目前,致病疫霉遗传转化体系已日趋成熟,农杆菌介导的转化技术具有操作便利、转化率高等特点,基于该转化技术编辑特定位点或构建突变体库无疑将大大方便后续研究,我们期待相关技术的进一步推广,推进后续的晚疫病研究。
基于测序技术和生物信息学分析方法展开的多组学研究,极大地推动了植物病理学发展。以高质量基因组为基础,利用多种测序和生物信息学分析方法,进行多组学联合分析,有助于发现关键位点或解析隐藏作用机制,为致病疫霉研究提供了巨大的助力。近年来,更多高质量基因组组装及注释结果被陆续发布,2022公布了第一个染色体级别的致病疫霉基因组全长为246.5 Mb,其中219 Mb的区域被映射到15条染色体上;研究者在全基因组范围内预测到19 981 个蛋白质,超过之前的预测结果;另外,该基因组中约72%的区域被注释为重复序列。通过与染色体级别高质量基因组对比,研究者发现致病疫霉有性生殖后代频繁出现染色体多倍体化或非整倍化现象,且不同染色体的发生频率存在差异,这一现象影响了基因表达,导致后代的生活力和致病能力发生多样的变化。此外,作者对介导甲霜灵抗性的致病疫霉基因进行了定位,确认决定抗药性的主要位点位于3号染色体上;这一区域频繁发生基因重组与染色体变异,产生了生长速度和抗药性水平多样的后代[222],可见高质量基因组对后续研究意义重大。Knaus 等发现致病疫霉基因组普遍存在拷贝数变异,大量菌株在特定位点上表现为三倍体甚至四倍体,且这种变异可以发生在保守的核心直系同源基因中,并不遵循双速基因组模型[223],这一现象的成因及后果还有待更多研究揭示。另外,细胞质基因组学研究也引起了研究者的关注,例如Shen 等对致病疫霉线粒体基因组的分析表明,不同地区的菌株间线粒体基因组存在极大的遗传分化,并可能为致病疫霉适应不同温度环境做出贡献[224]。多组学研究在植物病理学工作中的地位,如今已不容忽视。目前,大量田间致病疫霉菌株的收集积累、致病疫霉完整高质量基因组的公布以及生物信息学技术的发展,为全面开展正向遗传学研究奠定了基础。整合、汇总不同田间菌株和诱变菌株的基因组信息,构建致病疫霉泛基因组,从中鉴定决定致病力的关键基因,建立泛功能基因组学、泛效应分子组学数据库,对于深入的毒力机制研究、进一步发掘新抗病资源、合理规划防治方案具有重要意义。
值得期待的是,多组学的发展可与人工智能领域的进展接轨。基于人工智能和机器学习技术,对蛋白质空间结构、互作关键位点进行预测,甚至根据靶标从头设计药物已成为现实[225],而相关技术需要大量组学数据作为基础。基于多组学研究,利用人工智能和机器学习技术,根据致病疫霉代谢和侵染过程中的关键靶标设计杀菌剂,从理论上已具有实现的可能。我们期待多组学研究思路将在未来的晚疫病研究中扮演更重要的角色。
在基础理论研究的基础上,田间预测预报手段的更新对于晚疫病防治也有积极意义。以高光谱成像技术、机器视觉技术为基础实现的马铃薯晚疫病快速检测已进入实际应用环节,以便携式检测仪实现晚疫病快速识别、分级和统计处理,大大方便了田间检测[226-229]。高光谱成像技术(hyperspectral imaging technology)和机器学习(machine learning)结合,可以实现田间复杂环境、多病害环境下的快速识别和监测[230]。环介导等温扩增技术(loop-mediated isothermal amplification, LAMP)可以应用于晚疫病的早期检测,且具有高灵敏性,有助于解决晚疫病发病初期症状不明显这一问题[231-232];侯冰茹等利用光谱技术检测马铃薯发病叶片的过氧化物酶(peroxidase)活性,经过数据拟合建立了基于过氧化物酶活性的马铃薯晚疫病患病成都预测模型,能有效评估田间晚疫病发病情况,进行病害分级[233]。从比利时引进的CARAH晚疫病预测模型,以及一系列我国自主研发的新预测模型已进入推广应用阶段,这些模型基于气象数据进行发病情况预测,在许多地区表现良好,田间自动监测仪收集的温度、湿度、降水量等数据为监测模型提供了足够精密的输入数据[234-237],在此基础上,基于物联网技术的晚疫病实时监测预警系统已经成为可能[238],有望实现预测预报和自动化防治手段的有机结合,实现自动、智能、低成本的晚疫病防治。
