基于宏基因组的两种白芨根际微生物群落结构研究
2023-10-09姜晓斌余碧霞王恒生
姜晓斌, 余碧霞, 刘 冬, 王恒生, 李 萍
(1.安庆职业技术学院 农业产业化集成技术协同创新中心, 安徽 安庆 246003;2.合肥师范学院 生物与食品工程学院, 安徽 合肥 230601)
土壤微生物主要由土壤细菌、古生菌和真菌组成, 广泛分布于根际和非根际土壤中, 其多样性和丰富度对调节有机质分解、土壤碳动态和养分循环等生态系统功能起着关键作用[1]. 植物根际作为植物与土壤生态系统物质交换的媒介[2], 根际微生物对植物的生长发育至关重要, 可提高植物生产力和抗逆性[3-5], 同时对植物的代谢物也具有较强的影响[6].
白芨〔Bletilla striata(Thunb.) Reichb.f.〕为兰科多年生草本植物, 按花色可分为黄花白芨(Bletilla ochracea)和紫花白芨(Bletilla striata). 白芨在我国已有2 000多年的药用历史, 主要产自贵州、四川、湖南、湖北、安徽、河南、浙江和陕西等省[7]. 据《中华人民共和国药典》记载, 白芨具有收敛止血、消肿生肌等功效, 主治咯血、吐血、外伤出血, 疮疡肿痛, 皮肤皲裂[8]. 这些疗效与其化学成分密切相关, 其活性成分主要为多糖、联苯、二氢菲、菲类、黄酮类、苹果酸2-异丁酯葡萄糖、氧苄酯、多酚等[9-10]. 研究发现, 白芨根际微生物和种植的外界环境可能影响其活性物质种类和含量[11-12]. 因此, 探究不同白芨根际土壤微生物结构特征, 对深入理解白芨与微生物间的相互关系提供了理论依据.
1 材料与方法
1.1 采样区域
两种白芨样品采自安庆职业技术学院白芨试验基地, 取5月份自然生长且长势一致的白芨.
1.2 取样方法与样品处理
2022年5月, 在白芨试验田10 m×10 m取样区域, 选择5块面积为1m×1m的样方进行随机取样. 采用五点取样法在样方内选择黄花白芨(BOS)和紫花白芨样品(BSS)进行采集, 取样范围为白芨植株根际周围5 cm, 土层深度为10 cm的根际土壤. 样品过2 mm筛, 放置于无菌离心管中(50 mL), 液氮保存, 用于土壤DNA的提取、宏基因组测序和土壤理化性质的测定.
1.3 土壤理化性质的测定
土壤样品预处理参考吴昊等报道的方法[13]进行. 土壤含水量使用JK-100F土壤水分仪测定, 土壤含碳量(TC)通过CE-440型元素分析仪测定, 土壤含氮量(TN)采用凯氏定氮法测定, 土壤pH和电导率(水土比浸提液为5∶1)使用Bante902P pH/电导率/TDS/盐度计测定.
1.4 宏基因组数据分析
土壤宏基因组DNA提取采用郭辉等报道的方法[14]进行, 使用凝胶纯化试剂盒纯化. 检测浓度满足要求后, 委托华大基因通过BGISEQ-500测序平台进行宏基因组高通量测序. 利用LEfSe对白芨样品的微生物宏基因组原始数量进行筛选, 使用Megahit软件(默认参数)进行宏基因组组装. 使用Metaquast软件对组装结果进行效果评估, 比较组装结果和参考序列, 获得组装序列的高质量Contigs数、最长Contig、N50等信息. 利用Kraken v2软件将序列与已构建的物种组成数据库进行比较分析. 使用R包中的Vegan软件来分析Alpha多样性指数和PCA[15].
2 结果与分析
2.1 两种白芨土壤理化指标差异分析
分别对两种白芨土壤样品的5项主要指标(全碳、全氮、含水率、pH值和电导率)进行评价和分析, 结果如表1所示. BOS组和BSS组土壤理化指标除全碳外, 均无显著性差异, 两组样品理化指标基本一致.

表1 黄花和紫花白芨根际土壤理化指标
2.2 两种白芨根际微生物组成
宏基因组测序结果显示, 单个样品测序量超过10 G, 平均序列数量达到3 600万, 表明微生物测序量充分且波动较小. 如表2所示, BOS和BSS微生物种群分类结果中, 53.01%和59.94%的序列为未知物种. 两种白芨根际中微生物序列占比分别为41.16%和33.32%, BOS组略高于BSS组. 白芨主要微生物相对丰度显示, 不同分类学水平上的微生物重复度呈现较高水平, 其中以细菌含量最多, 分别为33.67%和25.66%, 其次是为真核生物, 分别为1.33%和1.29%, 再次为病毒, 分别为0.15%和0.16%.

表2 黄花和紫花白芨根际微生物测序结果
在门水平上共检测到56个菌门, 其中变形菌门(Proteobacteria)、放线菌门(Antinobacteria)、浮霉菌门(Planctomycetes)含量占比较高, 三者在白芨样品中的平均含量都超过75%. 在属水平上共检测到1 666个菌属, 丰度最高的均为链霉菌属(Streptomyces), 它在BOS和BSS样品中的平均含量分别为37.5%和26.2%. 其次是假单胞菌属(Pseudomonas), 第三是伯克霍尔德菌属(Burkholderia). 另外, 由图1可知, 在BOS和BSS样本间的链霉菌属和假单胞菌属丰度均存在显著性差异(p< 0.05).
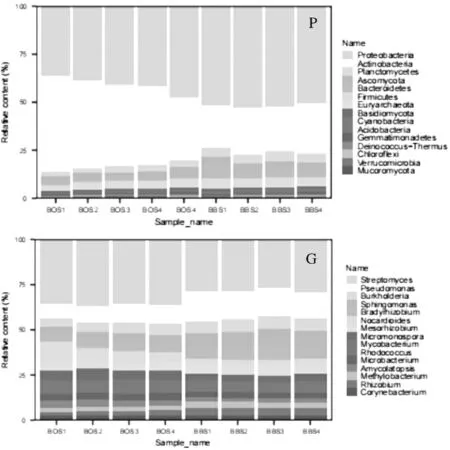
图1 门(P)和属(G)微生物组成
2.3 两种白芨样品微生物α多样性分析
在生态学中, 香农指数(Shannon)和Chao 1指数常用来估计物种总数[16]. 图2结果显示, 两种白芨根际微生物存在显著的差异性. 紫花白芨的Shannon和Chao 1指数均高于黄花白芨, 表明紫花白芨根际微生物的多样性更加丰富.
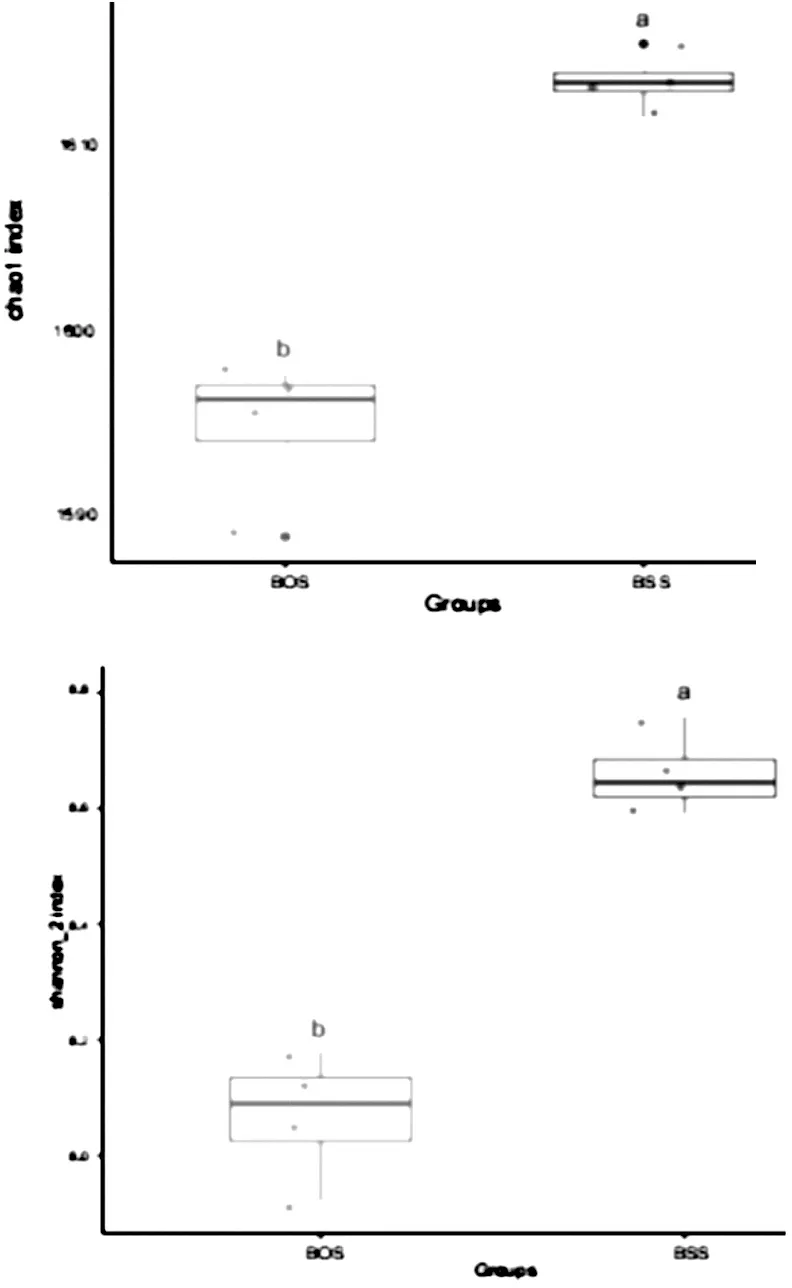
图2 两种白芨样品根际微生物多样性
2.4 两种白芨样品微生物主成分分析
进一步对白芨样本相似度进行分析, 结果如图3所示. 由图3可知, 同一组的样本距离较近, 说明组内样品均匀. 在属水平上, PC1、PC2和PC3的总体解释度达98.6%.
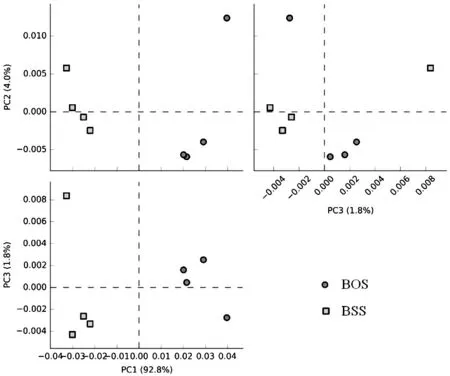
图3 两种白芨根际微生物组成主成分分析
2.5 两种白芨根际微生物分析
对两种白芨根际微生物进行分类, 结果显示, 差异水平从门到种均存在. 组间差异显著性检验结果表明(见图4a), 在属水平上有29个菌属存在显著性差异(P<0.05), 主要包括链霉菌属、类诺卡氏菌属(Nocardioides)、假单胞菌属和鞘脂菌属(Sphingobium)等菌属;在种水平上有33个菌属(见图4b)存在显著性差异(P<0.05), 主要包括威尼斯链霉菌(Streptomyces venezuelae)、伍氏束缚菌(Conexibacter woesei)、沼泽红假单胞菌(Rhodopseudomonas palustris)、印度鞘氨醇单胞菌(Sphingomonas indica)和黄克里伯菌(Kribbella flavida)等菌属.
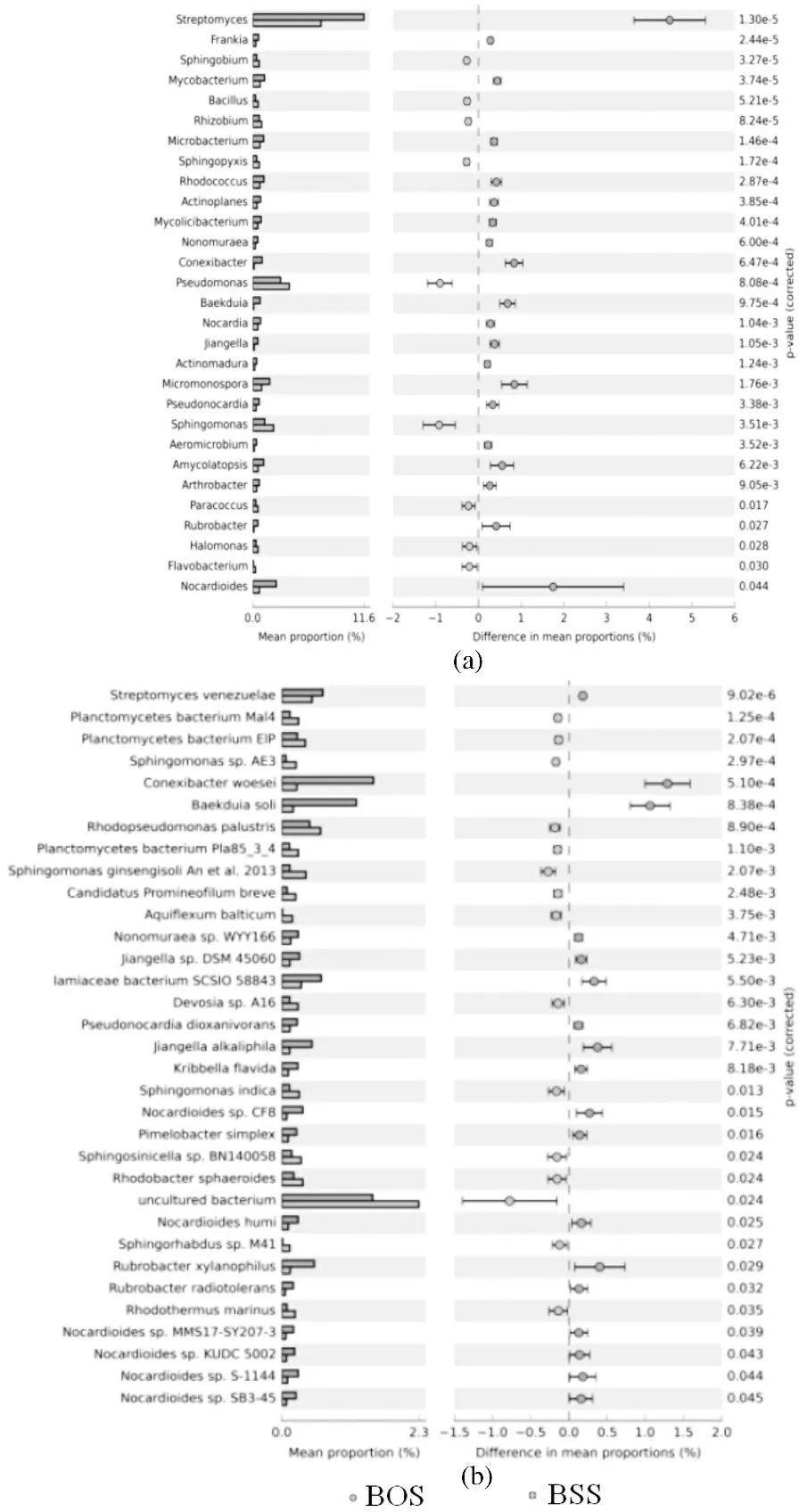
图4 两种白芨根际微生物差异情况
2.6 两种白芨根际土壤微生物含量相关性分析
为研究两种白芨根际土壤微生物丰度间的相互关系, 我们对两组丰度排名前17的微生物进行相关性分析, 结果如图5所示, 它们分别为棒状杆菌属(Corynebacterium)、分枝菌酸杆形菌属(Mycolicibacterium)、分枝杆菌属(Mycobacterium)、游动放线菌属(Actinoplanes)、根瘤菌属(Rhizobium)、中慢生根瘤菌属(Mesorhizobium)、链霉菌属、类诺卡氏菌属、红球菌属(Rhodococcus)、微杆菌属(Microbacterium)、拟无枝菌酸菌属(Amycolatopsis)、诺卡菌属( Nocardia)、假单胞菌属、鞘脂菌属、伯克霍尔德氏菌属 、贪铜菌属(Cupriavidus)和堆囊菌属(Sorangium). 中慢生根瘤菌属与链霉菌属、类诺卡氏菌属、红球菌属、微杆菌属、诺卡菌属和分枝杆菌属呈现正相关关系, 且相关性较高. 分枝菌酸杆形菌属与12个属呈现正相关关系. 链霉菌属与中慢生根瘤菌属、棒状杆菌属和游动放线菌属等6个属呈现正相关关系, 与假单胞菌属和根瘤菌属呈现负相关关系, 这可能与链霉菌属产生的代谢产物抑制了假单胞菌属和根瘤菌属微生物的生长相关[17-18]. 假单胞菌属与9个属呈现负相关关系, 类似的报道在多种植物根际中也被观察到. 此外, 不同微生物丰度之间呈现出显著的正相关或负相关关系(p<0.05), 说明不同微生物间互作密切, 共同影响着白芨根际营养代谢过程. 土壤中微生物是由多种菌群构成, 各类菌群之间形成相互促进或抑制的关系是必然且合理的, 而这种竞争通常是对能源和物资的竞争, 被称为生态位竞争, 其最终目标是通过竞争或共生的手段使种群生态适应性最大化[19]. 通过分析不同微生物之间含量的相关性发现, 伯克霍尔德氏菌属的存在会促进纤维堆囊菌属、贪铜菌属含量增加. 这是因为伯克霍尔德氏菌是一种在促进植物生长、适应环境方面十分有效的微生物[20]. 该结论与Zang等[21]关于伯克霍尔德氏菌属对植物有益, 可以促进部分有益菌定殖的结论相一致, 所以认为在植物根际微生物菌群之间存在一定的促进关系是合理的. 链霉菌属是一类能产生重要抗生素的类群, 是重要的药用资源. 研究发现, 该属广泛分布于盐碱度较高的土壤和极端环境中[22], 这有利于改善和提高白芨根系发育以及对外界环境胁迫的适应性.
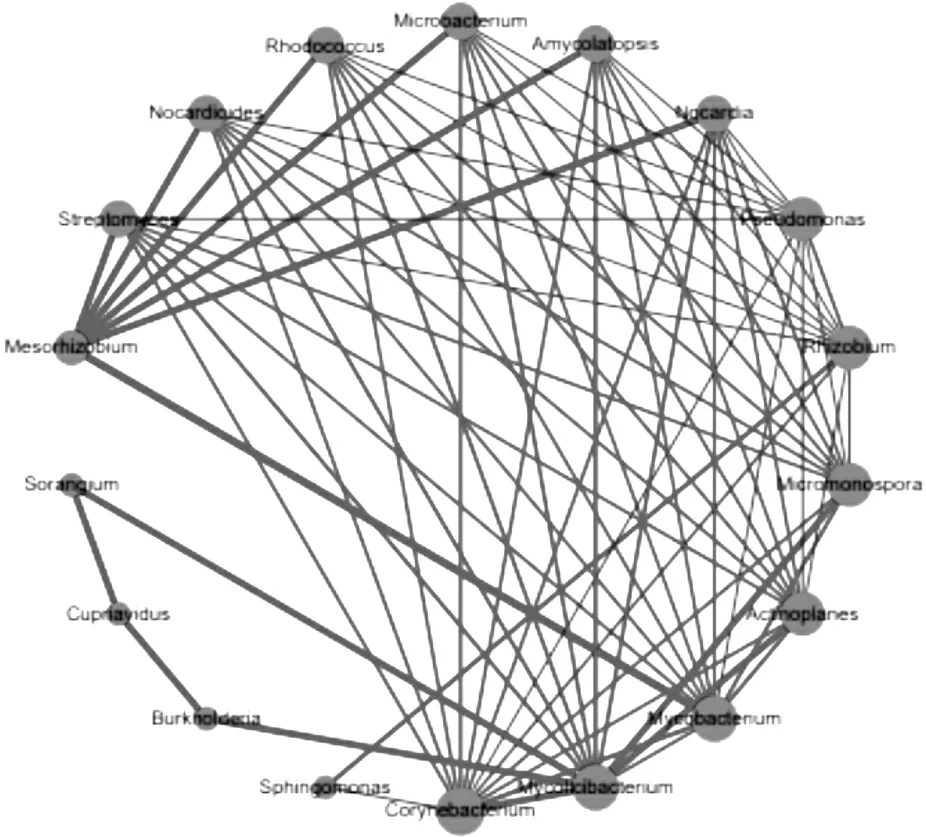
注释:绿点代表不同微生物, 点的大小代表其丰度的大小, 红色、蓝色连接线分别表示两种土壤微生物之间呈现正相关、负相关关系, 线的粗细程度代表两类菌相关性绝对值的大小.图5 根际与非根际土壤微生物相关性分析
3 结论
宏基因组测序结果显示, 两种白芨根际微生物菌群种类丰富, 紫花白芨根际微生物的物种丰富度比黄花白芨微生物物种丰度更高, 它们的微生物菌群差异水平从门到种均存在. 这些丰度较高的根际微生物存在正相关或负相关性.
