六价硫氟交换化学在药物设计中的应用*
2023-09-27桂馨墨秦艺曼
张 煚,桂馨墨,秦艺曼
(炎症免疫性疾病安徽省实验室 安徽医科大学药学院,安徽 合肥 230032)
磺酰氟类化合物的合成及性质的研究最早可以追溯到20世纪20年代的德国,随后,这些磺酰氟化合物被尝试作为酶抑制剂。Gold和Fahrney在1963年首次介绍了磺酰氟对酯酶的抑制作用[1],并测定了不同结构的磺酰氟对乙酰胆碱酯酶、α-胰凝乳蛋白酶和胰蛋白酶的反应速率。20世纪,关于磺酰氟类酶抑制剂的研究,产生了众多的成果。如今,细胞裂解液中为了防止蛋白降解而采用的蛋白酶抑制剂,如PMSF和AEBSF,即为磺酰氟类化合物。在早期的药物开发研究中,Baker是研究芳基磺酰氟类共价抑制剂的先驱。早在20世纪60年代,Baker等就使用芳基磺酰氟类化合物作为二氢叶酸还原酶、胰蛋白酶、α-胰凝乳蛋白酶、鸟嘌呤脱氢酶和黄嘌呤氧化酶等多种酶的不可逆共价抑制剂[2-7]。Colman团队亦在磺酰氟类共价抑制剂的研究中做了大量的工作[8-11]。但总的来说,长期以来,硫氟化合物在药物设计领域没有得到足够的认识,关注度较低。
2014年,Scripps研究所的Sharpless(2001和2022年两次诺贝尔化学奖得主)团队首次提出六价硫氟交换化学(Sulfur(VI)fluoride exchange,SuFEx)是一类新型的点击化学[12]。自此之后,六价硫氟交换化学引起了人们极大的关注,众多重要的研究成果不断涌现,已被广泛应用于有机合成、材料化学、化学生物学和药物化学等领域中[13]。鉴于六价硫氟交换化学在药物分子的设计合成中日益增加的重要性,本文介绍了近年来SuFEx在药物设计中的应用情况。
1 六价硫氟类化合物的结构及理化性质
目前,在药物设计中得到广泛应用的六价硫氟类化合物主要是烷基磺酰氟、芳基磺酰氟、芳基氧磺酰氟和芳基氟磺酰亚胺(图1A)。相对于磺酰氯,磺酰氟具有更为惰性的化学反应活性,能耐受还原性条件,在生理pH的水溶液中具有较长的半衰期,具有相对较好的生物相容性,和亲核试剂反应时只通过加成消除的取代机理形成共价键[14]。六价硫氟类弹头与靶蛋白中的亲核性氨基酸残基反应,能够形成稳定的共价复合物(图1B),但在特殊的情况下,芳基氧磺酰氟与赖氨酸形成的共价键也会因蛋白介导的水解作用水解为氨基磺酸[15]。

(A)六价硫氟类化合物的结构;(B)六价硫氟化合物与靶蛋白反应形成共价复合物。图1 六价硫氟类化合物
为了更好地设计含六价硫氟弹头的共价抑制剂,Mukherjee等详细考察了烷基磺酰氟、芳基磺酰氟、乙烯基磺酰氟、芳基氧磺酰氟和芳基氟磺酰亚胺等六价硫氟化合物(图1A)在生理条件下与亲核性氨基酸残基的反应活性[15],结果表明:烷基磺酰氟的反应活性最高,稳定性最差;乙烯基磺酰氟和芳基氟磺酰亚胺的稳定性高于芳基磺酰氟且活性与取代基相关;芳基氧磺酰氟最为惰性,反应活性最差,十分稳定;芳基磺酰氟的反应活性和生理条件下的水解性均与芳环上取代基直接相关,吸电子的取代基大幅提高活性的同时降低其水解稳定性。因此,在药物化学中,设计共价抑制剂时可以据此优化目标化合物的结构,调节反应活性与选择性,以达到恰当的平衡。
2 含六价硫氟官能团的小分子抑制剂
近年来,共价抑制剂的研究得到了蓬勃发展。与非共价药物相比,共价键的引入可以提高药物的活性,延长药物在体内的驻留时间等。对那些难以成药的靶点来说,共价药物是个很好的选择。在传统的共价药物设计时,通常借助于α,β-不饱和酰胺(酮)等结构[16],但共价药物的化学选择性,以及伴随而来的脱靶效应一直困扰着我们,迟滞了共价药物的开发。六价硫氟类(SuFEx)弹头,尤其是芳基氧磺酰氟,在生命体系中能够恰当地平衡反应活性和选择性。与靶标蛋白结合后,基于临近效应,能够高度选择性地与特定的氨基酸残基(Tyr、Ser、Thr 和Lys 等)反应,形成稳定的共价复合物;而与蛋白组的反应性很低,脱靶效应很弱。因此,含有SuFEx弹头的化合物具有作为共价药物的巨大潜力。几种典型的含六价硫氟官能团的小分子抑制剂案例如下文所示。
泛素蛋白酶体系统(UPS),是真核细胞内蛋白质降解的主要途径,负责降解错误折叠、损伤的非正常蛋白质,对维持细胞正常生理状态具有十分重要的作用。同时,它也是抗肿瘤靶点,目前已有硼替佐米、卡非佐米等蛋白酶体抑制剂被批准上市,用于治疗多发性骨髓瘤等恶性肿瘤。鉴于磺酰氟独特的化学性质,Brouwer等设计合成了一系列氨基酸磺酰氟衍生物[17-18],并基于此开发了选择性高且活性好的拟肽类磺酰氟蛋白酶体抑制剂(Peptido Sulfonyl Fluorides,PSFs)[19-24](图2)。化合物6对丝氨酸蛋白酶具有一定的抑制作用,其对糜蛋白酶的Ki值为 22 μmol/L。化合物7对酵母20S蛋白酶体的IC50达到 7 nmol/L。化合物8对蛋白酶体的抑制作用稍弱,但具有一定的选择性,对蛋白酶体β5i亚基的抑制作用比β5c亚基高25倍。进一步的修饰发现,具有游离氨基的拟肽类蛋白酶体抑制剂9-11具有纳摩尔级的IC50值,并且在不同的蛋白酶体亚基间具有显著的选择性。

图2 拟肽类磺酰氟抑制剂的结构及活性
转甲状腺蛋白(transthyretin,TTR),一种以四聚体形式转运甲状腺素和视黄醇的重要蛋白。在病理情况下,其解聚为单体,并错误折叠或组装生成淀粉样纤维,从而导致多种淀粉样蛋白集聚相关的疾病。Kelly等合成了两个芳基磺酰氟(12,13)和两个芳基氧磺酰氟(14,15)取代的1,3,4-噁二唑类TTR共价稳定剂(图3)[24-25]。该类化合物可以共价地与甲状腺素结合口袋中Lys15共价结合,形成磺酰胺键,从而稳定TTR。其中,芳基氧磺酰氟类化合物14和15由于活性较低,与Lys15的反应较慢,且不能反应完全,但对TTR的选择性更高,具有较弱的脱靶效应。此外,其与TTR形成的共价物显示出很强的荧光性,可以用来在活细胞和秀丽隐杆线虫体内检测TTR。一个有趣的现象是,化合物14和15与TTR中Lys15形成的芳基氧磺酰胺共价物会因蛋白结构的诱导作用水解为TTR-SO3。

图3 六价硫氟类TTR共价稳定剂
抗生素耐药是当前严重威胁人类健康的因素之一,为了克服细菌的多药耐药,笔者在Sharpless教授指导下基于SuFEx化学,由酚或酚的前体制备芳基氧磺酰氟衍生物,通过抗菌活性的表型筛选,发现白藜芦醇、己雷琐辛与和厚朴酚的芳基氧磺酰氟衍生物(16-18)(图4)可以有效杀死MRSA、VRSA、VRE和多种临床分离得到的多药耐药菌。这些衍生物没有明显的细胞毒性,能够高效破坏细菌的生物膜且不易产生耐药菌株,己雷琐辛的衍生物还与抗菌药物链霉素和庆大霉素具有协同效应[26]。

图4 具有抗耐药菌活性的白藜芦醇、己雷琐辛与和厚朴酚的芳基氧磺酰氟衍生物
常规的药物发现策略是通过筛选或合理药物设计来获得靶向特定蛋白的小分子。Kelly团队开发了一种逆向药物发现方法学,即通过温和反应性的小分子来识别蛋白质组中的亲核位点,再进一步有针对性地优化先导物获得潜在药物分子。SuFEx类化合物在生物体内的反应性和稳定性间具有良好的平衡,适合在逆向药物发现策略中作为探针。Kelly团队制备了芳基氟磺酰亚胺类小分子化合物库,并进一步鉴定这些SuFEx化合物所共价结合的蛋白质。其中,基于胸苷分子的芳基氟磺酰亚胺19(图5)可以共价结合多聚ADP核糖转移酶1(poly ADP-ribose polymerase 1,PARP1)并抑制其活性,且在活细胞中也具有良好的活性。基于此开发的药物分子有潜力作为治疗癌症的PARP1非共价抑制剂的有效补充[27]。

(A)芳基氟磺酰亚胺类衍生物的合成;(B)基于胸苷分子的芳基氟磺酰亚胺衍生物19。图5 逆向药物发现策略考察芳基氟磺酰亚胺类的作用靶点
3 基于SuFEx的化合物库构建
SuFEx类官能团的化学稳定性高,但在合适的反应条件下针对特定官能团又具有十分高效的反应活性,选择性高,因此可以用来模块化地构建衍生物库,以供生物活性筛选。例如,很多临床使用的药物分子中含有酚羟基,而硫酰氟(SO2F2)可以高度选择性地修饰酚羟基。Liu等采用后期官能团化(Late-Stage Functionalization,LSF)的衍生物合成策略,用SO2F2气体的乙腈溶液在96孔板中一次性将39个含酚羟基的抗癌药物原位转化为芳基氧磺酰氟衍生物,测试发现相对母体化合物,氧磺酰氟标记的衍生物20F-22F(图6)可以大幅提高抗癌活性[28]。

图6 芳基氧磺酰氟类抗癌药物
针对苗头化合物进行结构优化,最终获得药物分子的过程十分漫长且充满挑战性。SuFEx点击化学具有生物相容性,基于此构建的衍生物库可以直接用于原位的生物活性筛选。Wolan团队和Sharpless团队合作报道了一种基于SuFEx点击化学快速制备芳基氟磺酰亚胺衍生物库的策略[29];研究者从细菌半胱氨酸蛋白酶SpeB的中等活性抑制剂(23,Ki=8 μmol/L)出发,通过SuFEx点击化学策略在微孔板中快速(过夜)合成460种衍生物(图7A),通过直接原位筛选发现了多个活性得以大幅提高的衍生物。其中,活性最高的衍生物Ki为 18 nmol/L,衍生物24活性较好(Ki为 67 nmol/L),水溶性好,细胞毒性低,代谢稳定性高,成药性好(图7B)。
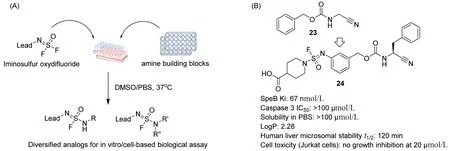
图7 基于SuFEx点击化学构建衍生物库发现细菌半胱氨酸蛋白酶SpeB抑制剂
4 基于SuFEx的共价蛋白药物

图8 非天然氨基酸FSY的结构及SuFEx介导的蛋白质偶联
5 基于18F/19F同位素交换的PET示踪剂的合成
基于18F的正电子发射断层显像(Positron Emission Tomography,PET)技术在疾病的诊断和病理研究等方面具有十分广泛的应用。未来这一技术的主要研究方向之一是在18F脱氧葡糖(18F-FDG)等示踪剂之外,开发更多具有靶向性的18F标记的示踪剂,以满足日益增长的精准医疗的需求。由于18F的半衰期较短,只有 110 min,因此在示踪剂中通过常见的碳-氟键引入18F,并在有限时间里快速分离纯化,非常具有挑战性。Scripps研究所Wu Peng团队证实了芳基氧磺酰酯在室温下可以快速与无机氟盐发生18F/19F同位素交换,并基于此开发了一种基于硫氟交换的超快18F标记技术[35]。这一技术得到的18F标记的示踪剂具有高比活度和高放化纯度。研究者应用该项技术在 2 min 之内便近乎当量地获得18F标记的PARP抑制剂奥拉帕尼,并且在小鼠的PET/CT中证实其对PARP高表达的肿瘤组织具有较高的选择性。基于该技术,He等合成了新型的阿尔茨海默病中β-淀粉样蛋白斑块(Aβ)的PET放射性示踪剂[36]。为进一步提高基于SuFEx的18F标记效率,Walter等开发了一种新的方案[37],即将[18F]F-负载到阴离子交换树脂上,并用BnEt3NCl的MeOH溶液洗脱,以获得18F标记的示踪剂。这种方法使得我们能够在高度稀释的溶液中对低纳摩尔量的芳基氧磺酰氟进行18F标记。和最初的方案相比,该方法避免了共沸干燥、碱的添加和HPLC纯化。作者利用这一方式,成功制备了29种18F-芳基氧磺酰氟,并且初步评估表明[18F]FS-DPA是一种非常有前景的易位蛋白(TSPO)表达可视化的示踪剂。Kim等[38]近来也开发了一种18F标记方案,即通过[18F]FSO2+转移试剂,可以原位地由苯酚或者胺制备芳基氧磺酰[18F]氟或氨基磺酰[18F]氟(图9)。

(A)18F标记的奥拉帕尼芳基氧磺酰氟衍生物;(B)18F标记的β-淀粉样蛋白斑块(Aβ)的PET示踪剂;(C)[18F]FS-DPA。图9 18F标记的PET示踪剂
6 展望
六价硫氟类化合物具有特殊的理化性质,在化学稳定性和反应性上具有很好的平衡。近年来,随着SuFEx的新型合成砌块、新型合成技术的不断涌现,药物化学家拥有了更多的工具来修饰小分子和生物大分子为六价硫氟衍生物,并基于此发现共价小分子和大分子药物。但当前六价硫氟化合物的反应机制尚未完全明确,该类共价抑制剂的脱靶效应,尤其是体内的毒副作用还需进一步研究。未来,随着研究的不断深入,六价硫氟交换化学在药物的设计领域具有广阔的应用前景。
