土壤铁氧化物–亚铁的相互作用及其环境影响研究进展①
2023-09-22余光辉
姚 远,余光辉,滕 辉*
土壤铁氧化物–亚铁的相互作用及其环境影响研究进展①
姚 远1,2,余光辉1,滕 辉1*
(1 天津大学地球系统科学学院,表层地球系统科学研究院,天津 300072;2 中国地质调查局西安地质调查中心,西安 710119)
铁氧化物和溶液相亚铁常在厌氧土壤环境中共存。铁氧化物能够加快亚铁的氧化速率,且控制亚铁氧化成矿产物的类型,同时,亚铁与铁氧化物组成的系统是一种良好的还原剂,能够有效还原重金属及降解有机污染物。另一方面,亚铁能够催化铁氧化物晶相转变,导致铁氧化物结构和表面性质发生改变,进而影响相关重金属、有机质的环境行为。本文综述了铁氧化物催化亚铁氧化成矿、铁氧化物–亚铁系统还原污染物以及亚铁催化铁氧化物相变的反应机制及影响因素,最后,对未来在自然土壤中研究铁氧化物–亚铁界面反应及其环境影响进行了展望。
铁氧化物;亚铁氧化;矿物相变;有机污染物;重金属
铁元素(Fe)是地壳中含量第四的元素,与其前后的Al和Ca金属元素不同, Fe元素具有更强的化学反应性,广泛参与环境中各类氧化还原反应,Fe的电子排布为[Ar]3d64s2,而[Ar]3d64s0和[Ar]3d54s0结构的稳定性确保Fe能以Fe(Ⅱ)和Fe(Ⅲ)的形式存在,同时,溶液相Fe3+转化为Fe2+的标准还原电位为0.771 V,这使得这类反应在自然条件下普遍发生。由于Fe元素含量丰富以及良好的氧化还原反应活性,Fe(Ⅱ) 的氧化与Fe(Ⅲ) 的还原驱动着环境众多元素的转化与物质循环,因此,研究Fe元素在环境中的氧化还原反应具有重要研究意义。
自然环境中,Fe主要以Fe(Ⅱ) 和Fe(Ⅲ) 形式存在,铁(氢)氧化物(文中统称为铁氧化物)是土壤中最活跃的组成成分之一[1],由于其良好的吸附能力及生物反应性,铁氧化物对土壤中重金属的钝化与固定、有机质的矿化与积累、养分元素的利用等众多环境过程有着重要影响[2]。Fe(Ⅱ) 相比Fe(Ⅲ) 更易溶解,导致Fe(Ⅱ) 在生物可利用的形式中丰度较高,Fe(Ⅱ) 可以通过岩石的物理、化学风化作用进入到环境中,也可以通过厌氧环境下化学还原剂(如硫化物)或异化铁还原菌还原Fe(Ⅲ) 矿物产生。这也使得溶液相Fe(Ⅱ) 常与铁氧化物共存,近二十年来,随着人们对Fe的氧化还原反应的研究,发现相比溶液相Fe(Ⅱ),与铁氧化物表面结合Fe(Ⅱ) 具有更强的还原反应性,例如:早在1999年Buerge和Hug[3]发现Fe(Ⅱ) 与铁氧化物组成的系统能够快速还原重金属Cr(Ⅵ)。之后围绕这一思路,一系列研究探索了厌氧体系中,Fe(Ⅱ) 与铁氧化物系统在还原降解众多重金属及有机污染物的反应,包括Cr(Ⅵ)、Hg(Ⅱ)、As(Ⅴ) 等重金属污染物[4]以及四氯化碳、硝基芳香烃、亚硝酸盐等有机污染物[5-10];并详细研究了这一系统反应性的影响因素,这类反应在土壤原位修复领域具有良好的应用前景。同时,在上述反应中,研究还发现铁氧化物表面具有模板效应,能够控制Fe(Ⅱ) 氧化成矿产物的类型。如Larese-Casanova等[11]发现反应中存在的针铁矿诱导Fe(Ⅱ) 氧化形成针铁矿,后续的研究表明在有氧环境下,这种模板效应依然存在,同时也发现铁氧化物的存在能够显著加快Fe(Ⅱ) 的氧化速率。另一方面,研究发现溶液相亚铁能够催化弱结晶态铁氧化物发生晶相转化形成结晶度高的铁氧化物。得益于斯堡尔光谱技术的应用,Williams 和Scherer[12]首先采用同位素标记及穆斯堡尔光谱的方法证实了Fe(Ⅱ) 与铁氧化物之间的电子传递作用,后续众多研究也报道了Fe(Ⅱ) 与铁氧化物之间的电子传递和原子交换现象[13-14]。Scherer等人的一系列研究表明Fe(Ⅱ)催化铁氧化物的晶体相变反应是由Fe(Ⅱ) 与铁氧化物之间的电子传递/原子交换作用驱动的[15]。基于众多界面电子传递和原子交换的研究,大多数学者认为亚铁催化铁氧化物相变的实质是电子传递和原子交换作用导致的铁氧化物固相转化或溶解重结晶,但目前关于铁氧化物如何发生溶解以及矿物如何成核生长仍缺乏直接证据,仍需持续研究关注。同时,在这一过程中,由于铁氧化物的矿物学的转变会导致其表面性质与反应活性的改变,进而影响与之共存的重金属、有机质等物质的环境行为,具有重要环境意义。
目前,铁氧化物–亚铁之间的相互作用机制及其环境影响已在室内控制实验中取得较好的进展,但实际土壤环境复杂多变,铁氧化物–亚铁之间的相互作用常处于动态变化之中,使得研究自然条件下铁氧化物–亚铁之间的界面反应变得更为困难,也导致预测与之相关的重金属、有机物等物质的环境行为面临十足的挑战。本文就铁氧化物–亚铁之间的相互作用以及相关环境影响的研究进行了总结,并以此为基础提出相关研究今后的重点和方向,进一步加快相关研究的发展和应用。
1 土壤常见铁氧化物类型及现代分析技术
铁氧化物普遍分布于各种类型的土壤中,由于表面反应活性高,在自然环境中,很难找到一个没有铁氧化物参与的反应[16]。目前,已报道的铁氧化物共16种[17],其中土壤中最常见的有水铁矿、纤铁矿、针铁矿、赤铁矿等。水铁矿(Fe(OH)3),经常分布在灰壤中的温和或冰冷的区域、酸性矿山废水污染区域及淹水水稻土中,具有比表面积大、结晶度差、表面反应活性高等特点,通常难以在环境中稳定持续存在,具有较高的标准吉布斯自由能((–469.9±1.2) kJ/mol)[18],易于转变为结构更稳定的铁矿物。常见的相变产物有磁铁矿、针铁矿、赤铁矿,因此水铁矿是一些热力学稳定铁氧化物的前驱体。此外,由于水铁矿粒径较小,且晶体结构复杂,目前关于水铁矿的结构尚未有统一的认识。纤铁矿(γ-FeOOH)常见于季节性交替氧化还原波动的土壤环境中,是一种亚稳态矿物,可以作为其他铁氧化物的前驱矿物[19],一般易于转化为针铁矿。纤铁矿结构为阴离子的立方密堆积(ccp)的伯姆石层状结构,属斜方晶系。针铁矿(α-FeOOH)几乎存在于所有类型的土壤、湖泊及河流沉积物中[20],通常是由黄铁矿或磁铁矿等其他铁矿物风化而来,属于斜方晶系,结构为立方体紧密堆积结构(ccp),一般与纤铁矿为同质多象,是一种热力学稳定性较高的铁氧化物,不易转变为其他铁氧化物。赤铁矿(Fe2O3) 普遍分布于热带、亚热带温度较高的土壤和沉积物中[17, 21]。赤铁矿具有六方阴离子紧密堆积(hcp)的刚玉结构,热力学稳定性强,通常不会转变为其他铁矿物相。在一定条件下,上述各类型铁氧化物晶体之间可以互相转化,在大多数自然条件下,由于吉布斯自由能差产生的驱动力,铁氧化物趋向形成热力学稳定的矿物(如针铁矿与赤铁矿),相关热力学参数见表1。
自然土壤环境多样进而导致土壤铁氧化物的化学结构与组成复杂,目前铁氧化物的矿物特征已经能够得到很好的表征认识(表2)。常规的传统表征技术手段如X射线粉晶衍射分析(XRD)、扫描电子显微镜(SEM)、透射扫描电子显微镜(TEM)、傅里叶变换红外光谱(FTIR)、X 射线光电子能谱(XPS)、动态光散射(DLS)、静态光散射(SLS)等能够较好地表征铁氧化物基本的组成、形貌、粒径、官能团以及分散与凝聚状态等特征,但由于上述技术的局限性,无法对矿物相关的晶体结构、价态、分子/原子的排列方式等特征进行更加深入的认识,限制了人们对铁氧化物及相关界面反应机理的探索。目前得益于穆斯堡尔谱(Mössbauer spectra)、原子力显微镜(AFM)、同步辐射X射线吸收光谱(XAS)、球差校正扫描透射电子显微镜(Cs-STEM)等一系列现代分析技术的快速发展,为人们了解铁氧化物及其相关界面反应提供了强大支持[24]。
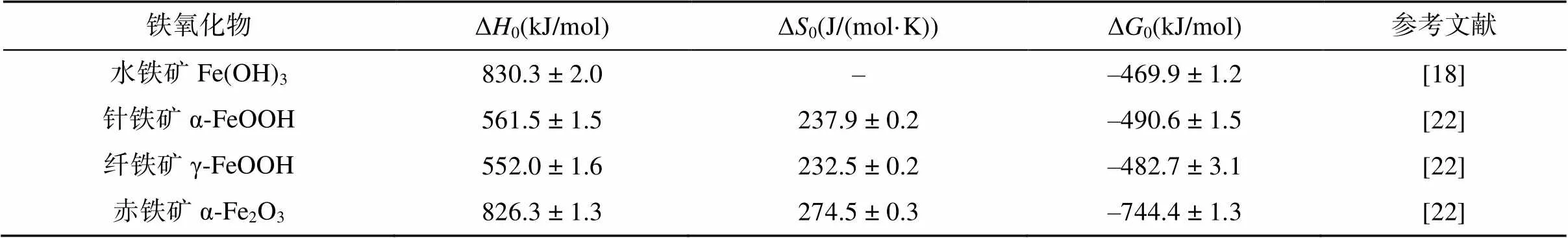
表1 土壤常见铁氧化物的热力学参数
注:焓(Δ)和吉布斯自由能(Δ)的参数条件为298 K、1 bar。

表2 研究矿物的现代分析技术汇总[23]
2 铁氧化物催化Fe(Ⅱ) 氧化成矿
Fe(Ⅱ) 的氧化包括非生物(化学)和生物过程[25],主要受控于自然环境中pH和O2浓度[26-27]。Fe(Ⅱ) 的非生物氧化成矿是土壤中次生铁氧化物形成的重要来源。一方面,亚铁的氧化可以通过溶液中的均相反应(homogeneous reactions)进行;另一方面,由于环境中存在各种矿物,因此亚铁的氧化经常在矿物影响下,以非均相反应(heterogeneous reactions)的方式进行。由于铁氧化物在土壤中大量存在,且反应活性高,因此许多研究关注在铁氧化物存在下,Fe(Ⅱ) 的氧化成矿过程。目前,众多研究发现铁氧化物表面可以催化Fe(Ⅱ) 氧化成矿,提高Fe(Ⅱ) 氧化速率,且明显控制新生铁氧化物类型与特征。
众多铁氧化物具有半导体的性质,能够促进矿物的电子传递过程[14],从而加快Fe(Ⅱ) 的氧化速率。同时,研究发现Fe(Ⅱ) 的氧化速率与铁氧化物结晶度以及吸附能力密切相关。Tamura 等人[28]研究了中性pH条件下,水铁矿、针铁矿和纤铁矿对Fe(Ⅱ) 氧化动力学的影响,发现在水铁矿存在时,Fe(Ⅱ) 的氧化速率大概是针铁矿、纤铁矿存在时的3倍,因此提出铁氧化物表面催化Fe(Ⅱ) 氧化的速率与其结晶度直接相关。而Jones等[29]研究发现在酸性条件下(pH 4.5 ~ 5.5),铁氧化物依然能显著加快Fe(Ⅱ) 的氧化速率,且Fe(Ⅱ) 的氧化速率正比于铁氧化物表面吸附态Fe(Ⅱ) 的浓度,由此认为铁氧化物催化Fe(Ⅱ) 氧化的速率是由铁氧化物对Fe(Ⅱ) 的吸附量所控制;类似地,Park和Dempsey[27]也报道了中性低氧条件下,水铁矿催化Fe(Ⅱ) 氧化速率受控于吸附态Fe(Ⅱ)的含量。然而,上述研究中反应体系限于纯反应系统,没有考虑自然土壤系统的多相性。近期Chen 和Thompson[30]研究了自然土壤环境下铁氧化物对Fe(Ⅱ) 的氧化成矿的影响,通过去除自然土壤中铁氧化物,与原土壤进行对比试验,发现土壤中铁氧化物的存在同样会加快Fe(Ⅱ) 的氧化速率。
此外,铁氧化物能够作为Fe(Ⅱ) 氧化成矿的模板,影响成矿产物的类型。通常,Fe(Ⅱ) 在溶液中的均相氧化会形成无定形Fe(Ⅲ)-OH络合物或亚稳态铁矿物(如水铁矿或纤铁矿)[31],而在结晶态铁氧化物存在下,Fe(Ⅱ)氧化会形成热力学稳定的铁氧化物(如针铁矿或赤铁矿)[6,11,31]。在这类反应中,Fe(Ⅱ) 在铁氧化物表面的反复吸附和氧化促使新的铁氧化物相的积累,导致新生铁氧化物相通常类似于吸附的铁氧化物表面。如:在厌氧体系中,Chun等人[6]在研究Fe(Ⅱ)-针铁矿系统还原降解4–氯硝基苯和三氯硝基甲烷的试验中,发现在针铁矿作用下,Fe(Ⅱ) 氧化后新形成的矿物也为针铁矿;同样,Larese-Casanova等人[11]发现针铁矿诱导Fe(Ⅱ) 形成针铁矿,而在磁铁矿与磁赤铁矿表面促使Fe(Ⅱ) 氧化形成纤铁矿。另一方面,在有氧环境下,研究者发现针铁矿表面同样促使Fe(Ⅱ) 氧化形成结晶度高的针铁矿[31]。此外,Chen和Thompson[30]发现在复杂的自然土壤环境中,上述铁氧化物的模板效应依然存在,即Fe(Ⅱ) 氧化产物类型强烈受控于土壤中预先存在的铁氧化物,由此可见铁氧化物对Fe(Ⅱ) 氧化产物类型的控制作用在自然环境中普遍发生。此项研究将先前类似的研究扩展至复杂的自然土壤环境,后续研究可在此基础上继续探索自然土壤环境中铁氧化物对Fe(Ⅱ) 的氧化成矿的影响。同时,当前的研究仅限于纯的铁氧化物反应体系,在土壤中,铁氧化物在形成过程中晶格常掺杂各种离子或分子杂质或者发生阳离子同晶替代,这些现象都会改变原来铁氧化物的结晶度、形貌、比表面积等矿物学特征以及表面反应活性。这些性质的改变是否会改变铁氧化物催化Fe(Ⅱ) 氧化的反应动力学及其对产物类型的控制,值得后续深入研究。
3 铁氧化物–Fe(Ⅱ)还原污染物及其反应性的影响因素
3.1 铁氧化物–Fe(Ⅱ)还原污染物
目前众多研究报道了铁氧化物与溶液相亚铁组成的系统还原降解众多污染物,包括四氯化碳、硝基芳香烃、亚硝酸盐等污染物[5-10]以及Cr(Ⅵ)、Hg(Ⅱ)、As(Ⅴ) 等重金属污染物[3-4](表3)。在这些还原污染物的反应中,无论使用何种类型的铁氧化物,相同污染物的反应产物都是相似的。其中有机污染物中的脱卤反应普遍发生,例如四氯化碳(CCl4)脱氯后生成氯仿[5, 32]。
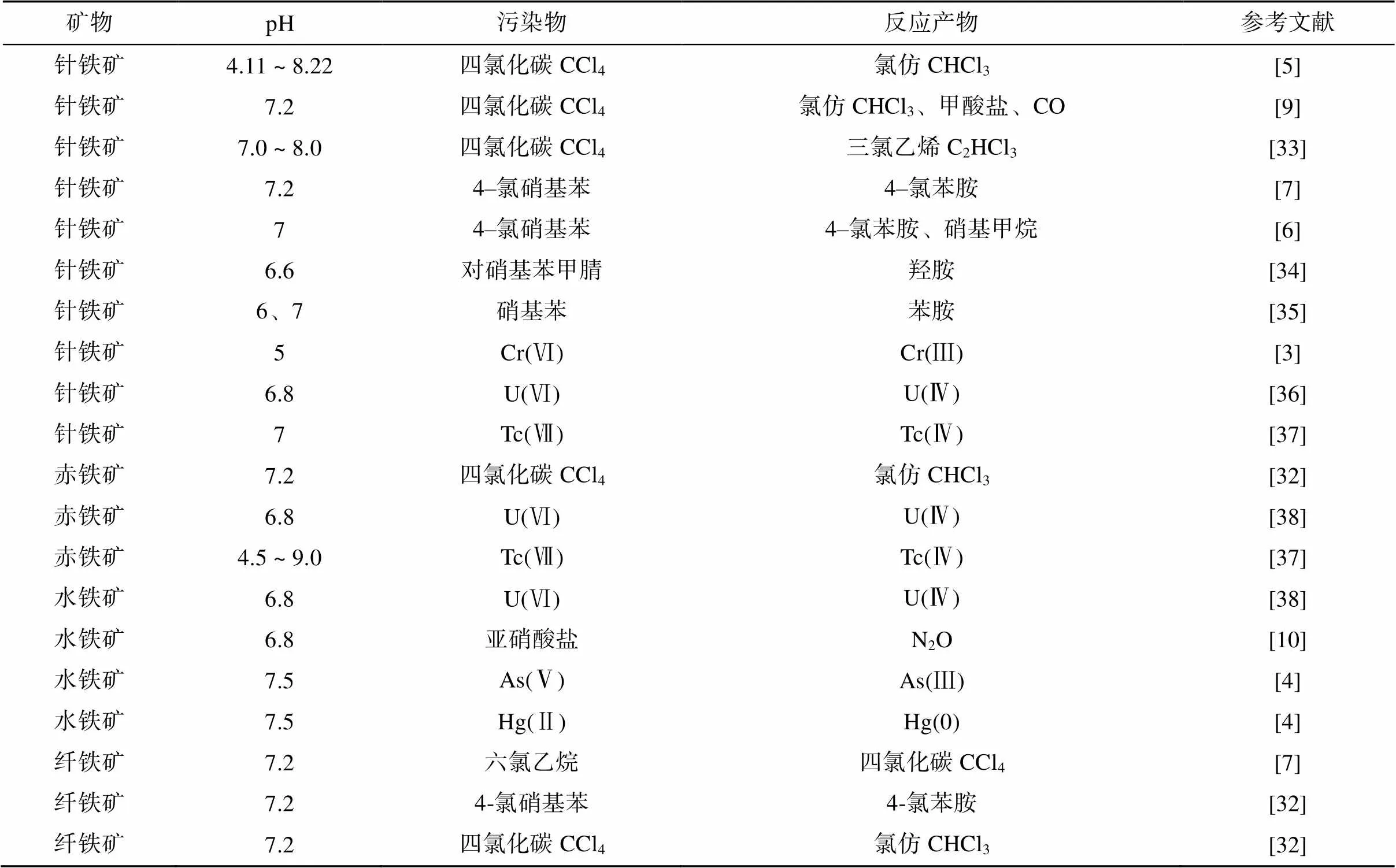
表3 铁氧化物–Fe(Ⅱ)系统还原污染物的相关报道
当前关于铁氧化物与亚铁组成的系统还原降解污染物的研究尚局限于单一的反应过程,缺少环境中多组分参与耦合反应的研究,后续研究需要考虑其他可能参与反应的物质,以模拟实际土壤中该类反应的发生。此外,在一些受污染土壤中存在有机无机复合污染的情况,该方面的研究目前较为薄弱,未来值得进一步关注。
3.2 铁氧化物–Fe(Ⅱ) 还原反应性的影响因素
在铁氧化物–Fe(Ⅱ)系统还原降解污染物的研究中,即使相同类型的铁氧化物和相同的污染物,吸附在铁氧化物表面上的Fe(Ⅱ) 的还原反应性也存在较大差异。该还原反应性与铁氧化物的聚集状态、暴露晶面等矿物性质以及反应pH、Fe(Ⅱ) 浓度、有机质的存在等反应条件密切相关。
铁氧化物颗粒的聚集会导致吸附在其表面的Fe(Ⅱ) 的还原反应性降低。例如,Amonette等人[5]研究发现,针铁矿颗粒的聚集导致其表面可用位点减少从而使得四氯化碳的降解速率降低。常用的有机缓冲液,如 MOP、HEPES等可增强铁氧化物纳米颗粒的聚集状态并降低这些系统的还原反应性[47]。另一方面,相比大颗粒铁氧化物,粒径较小的铁氧化物由于更大反应比表面积以及更高的表面反应能,通常具有更强的氧化还原反应性。然而,Cwiertny等人[48]在研究纳米针铁矿与亚铁组成的系统还原硝基苯的反应中,发现不同粒径的纳米针铁矿都发生显著的聚集,形成微米级的密集聚集体,当还原速率以比表面积归一化后,却发现粒径小的针铁矿纳米颗粒系统还原反应性低于较大粒径针铁矿的还原反应性。因此,直接使用比表面积来评估聚集颗粒悬浮液中表面反应性的差异并不合理,为了真实地评估这类反应中的纳米级尺寸效应,需要开发新的方法来量化湿纳米颗粒在悬浮液中可用于吸附和反应的表面积量。
铁氧化物的暴露晶面能够影响吸附态Fe(Ⅱ)的还原反应性。Chun 等人[6]研究发现针铁矿{021}晶面损失,会降低Fe(Ⅱ)–针铁矿系统对4–氯硝基苯和三氯硝基甲烷还原反应性;Huang等人[21]研究表明,赤铁矿{110}晶面可以将Fe(Ⅱ) 限制在五配位排列中,而{001}晶面可以将Fe(Ⅱ) 限制为六配位排列,相比在赤铁矿{001}面上的六配位排列,赤铁矿{110}面上的五配位排列表现出更强的Fe(Ⅱ)吸附能力。更重要的是,赤铁矿{110}晶面上Fe(Ⅱ)的五配位排列方式比赤铁矿{001}面上的六配位排列能更加有效地催化过氧化氢转化为羟基自由基,从而降解各种有机污染物。因此,铁氧化物的晶面特异反应性对于环境修复具有广泛的意义。目前部分研究聚焦开发具有优势晶面的铁氧化物颗粒,以提高 Fe(Ⅱ)–铁氧化物系统还原降解重金属及有机污染物的能力,未来该方面的研究值得持续关注。
pH通过影响Fe(Ⅱ) 在铁氧化物表面的吸附行为,从而影响Fe(Ⅱ)–铁氧化物系统的还原反应性。自然环境中pH范围一般为5 ~ 7.5,当 pH<5时,矿物表面吸附的Fe(Ⅱ) 的量非常有限,导致还原反应性低;而pH>7.5时,则会形成Fe(Ⅱ) 沉淀物。在自然pH范围内,随着 pH 的增加,矿物对Fe(Ⅱ) 的吸附变强,从而提高还原反应性。另一方面,pH会影响吸附于矿物表面的Fe(Ⅱ) 类型。Zhang等人[49]研究了Fe(Ⅱ) 在纤铁矿上的吸附,发现在纤铁矿表面形成两类Fe(Ⅱ) 表面络合物,其中,Fe(Ⅱ)单羟基表面配合物 (≡FeⅢOFeⅡOH-)是主要类型,而(≡FeⅢOFeⅡ)+是次要类型。后来研究采用表面络合模型(surface complexation model) 拟合Fe(Ⅱ) 在氧化铁上的吸附的研究[50](图1),方程(1)和(2)显示了这两种稳定的表面Fe(Ⅱ)络合物的形成。
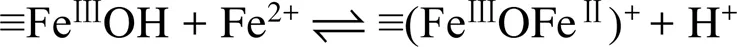
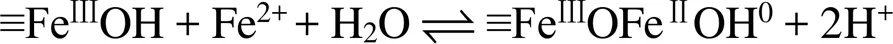
这两种形式的Fe(Ⅱ) 依赖于溶液pH,且研究表明4–氯硝基苯、U(Ⅵ) 还原速率与以(≡FeⅢOFeⅡOH-)形式存在的吸附态Fe(Ⅱ) 浓度呈正比关系[50-51]。然而,随后的研究发现吸附态Fe(Ⅱ) 与铁氧化物结构Fe(Ⅲ) 发生电子传递作用,尤其在低Fe(Ⅱ) 浓度条件下,吸附态Fe(Ⅱ) 的存在是瞬时的,Fe(Ⅱ) 会迅速与铁氧化物结构Fe(Ⅲ)发生界面电子传递并产生新的铁氧化物[12, 43],说明表面络合模型中所提到的Fe(Ⅱ) 表面络合物并不会在反应中稳定存在,因此,该模型在这类反应中的适用性需进一步研究讨论。
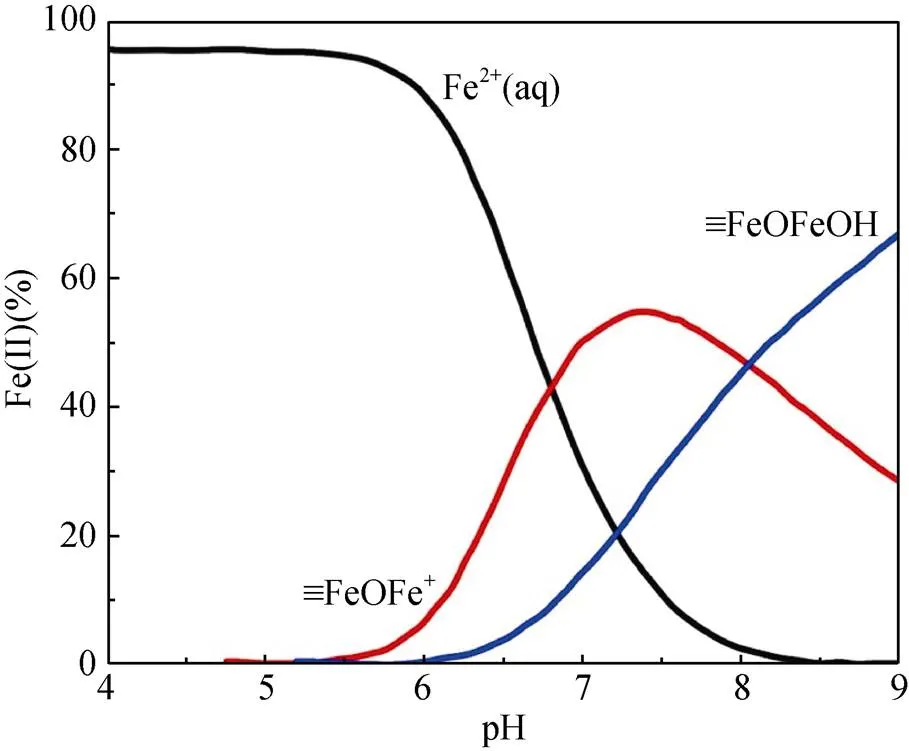
图1 吸附于赤铁矿表面的Fe(Ⅱ) 类型的分布[24]
溶液相Fe(Ⅱ) 的浓度也是影响矿物表面Fe(Ⅱ) 还原反应性的重要因素。在低Fe(Ⅱ) 浓度下,Fe(Ⅱ) 浓度的增加会提升铁氧化物对Fe(Ⅱ) 的吸附量,这促进了水溶液 Fe(Ⅱ) 和晶格Fe(Ⅲ) 之间的界面电子传递,并产生更快的还原反应。在高Fe(Ⅱ) 浓度下,尤其是吸附的 Fe(Ⅱ) 量超过了铁氧化物表面单层吸附位覆盖量,导致界面电子传递速率快速降低,因此氧化还原反应性受到抑制[15]。
有机质(如富里酸、胡敏酸、胡敏素等)可以通过静电作用、氢键作用、配体交换和疏水作用吸附在铁氧化物表面[52],从而影响吸附态Fe(Ⅱ) 的反应性。Colón等人[34]研究表明有机质主要通过络合吸附于针铁矿表面的Fe(Ⅱ),抑制吸附态Fe(Ⅱ) 与污染物之间的电子传递作用,从而降低Fe(Ⅱ)–针铁矿体系的还原反应性。同样地,Vindedahl等人[53]也发现一系列有机质的添加,包括Pahokee Peat 腐植酸、Elliot土壤腐植酸、Suwannee River腐植酸、Suwannee River NOM、Suwannee River富里酸I、Suwannee River富里酸Ⅱ和Pahokee Peat 富里酸,都在不同程度上抑制了Fe(Ⅱ)–针铁矿系统对4–氯硝基苯 (4-ClNB) 的还原反应性。同时在这些NOM-Fe(Ⅱ)–针铁矿体系中,发现随着有机质的分子量和氮、碳和芳烃含量的增加,4-ClNB的还原降解速率更快,而随着羧基、氧、杂脂族和脂肪族含量的增加,还原速率更慢,这一发现为预测有机质与Fe(Ⅱ)、铁氧化物组成的系统的还原反应性提供了依据。
4 亚铁催化铁氧化物相变及其环境影响
环境中,各类型铁氧化物晶体之间可以互相转化,铁氧化物之间的晶相转化有溶解再结晶或直接固相转化两种方式。溶解再结晶是指弱结晶的铁氧化物先溶解,产生Fe(OH)2+和 Fe(OH)+等羟基铁离子,然后再通过聚合作用形成结晶度高的铁氧化物;固相直接转化机制是弱结晶铁氧化物通过脱水和结构重排直接转化为结晶度高的铁氧化物[54]。在有氧环境中,针铁矿和赤铁矿是两种热力学稳定的铁氧化物相,也是大多数铁氧化物转化的最终矿物相。土壤环境中,结晶度差的水铁矿一般作为晶体铁氧化物的前驱矿物,由于比表面积大及表面活性高,水铁矿对重金属等污染物具有很高吸附容量,因此研究水铁矿的相变具有重要环境意义,目前,亚铁催化水铁矿相变受到广泛关注。
4.1 亚铁催化铁矿物相变的反应机制
亚铁催化铁氧化物相变的实质是Fe(Ⅱ) 与铁氧化物之间的界面电子传递和原子交换作用,使得铁氧化物直接发生固相转化或溶解重结晶[15]。Williams 和Scherer[12]首先采用同位素标记及穆斯堡尔谱的方法证实了这一现象。这种界面电子传递作用与铁氧化物结构中一些晶格Fe(Ⅲ) 的还原溶解行为密切相关[14]。对此Handler等人[13]提出了Fe(Ⅱ) 与铁氧化物之间电子传递的模型,很好地揭示了Fe(Ⅱ) 与铁氧化物之间电子传递作用的具体反应过程 (图2)。近年来,Scherer课题组系统研究了溶液相Fe(Ⅱ) 催化各类型铁氧化物的晶体相变过程,结果也表明:铁氧化物结构态Fe(Ⅲ) 与Fe(Ⅱ) 发生原子交换,Fe(Ⅱ) 进入铁氧化物表面或结构中被氧化为Fe(Ⅲ),形成次生铁氧化物,而原始铁氧化物中结构态Fe(Ⅲ) 则被还原,发生溶解释放到溶液中去,重新成核结晶形成新的更加稳定的铁氧化物[15]。而Jones等人[55]研究发现吸附于铁氧化物表面的硅酸盐能够抑制Fe(Ⅱ) 的吸附,导致Fe(Ⅱ) 与矿物结构态Fe(Ⅲ) 之间的原子交换减少,从而抑制了Fe(Ⅱ) 催化水铁矿晶相转化过程;此外,还发现水铁矿晶格内的硅酸盐并没有降低Fe(Ⅱ) 与水铁矿之间的原子交换,但依然抑制了水铁矿的相变。这表明水铁矿的溶解过程释放出来的硅酸盐抑制了新矿物的结晶,由此也进一步支持了Fe(Ⅱ) 催化的Fe(Ⅲ) 矿物相变的溶解重结晶机制。
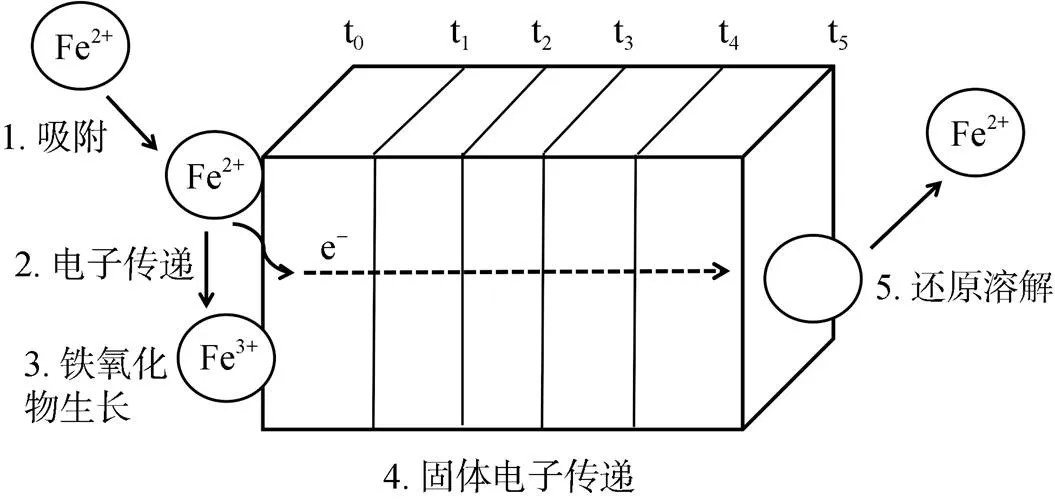
图2 亚铁与铁氧化物之间的电子传递机制模型(修改自Handler等[13])
此外,Boland等人[56]提出一种新的Fe(Ⅱ) 催化水铁矿相变过程的解释(图3):Fe(Ⅱ) 吸附到水铁矿表面后通过电子传递作用立即氧化成 Fe(Ⅲ),由此产生的电子传递到水铁矿晶格Fe(Ⅲ),将Fe(Ⅲ) 还原为Fe(Ⅱ) 释放到溶液中,氧化产生的Fe(Ⅲ) 会沉淀为活性水铁矿,其作为新矿物生长结晶的核,同时也能进一步吸收溶液中的Fe(Ⅱ),这种作用不断重复进行,直至活性水铁矿相变为针铁矿。目前,一系列铁氧化物与Fe(Ⅱ) 之间的电子传递及原子交换过程的发现,极大促进了人们对亚铁催化水铁矿相变的过程认识,但除了动力学以及晶格离子溶出现象,尚缺乏直接的实验证明水铁矿的实际晶相转变过程,未来仍需进一步研究。
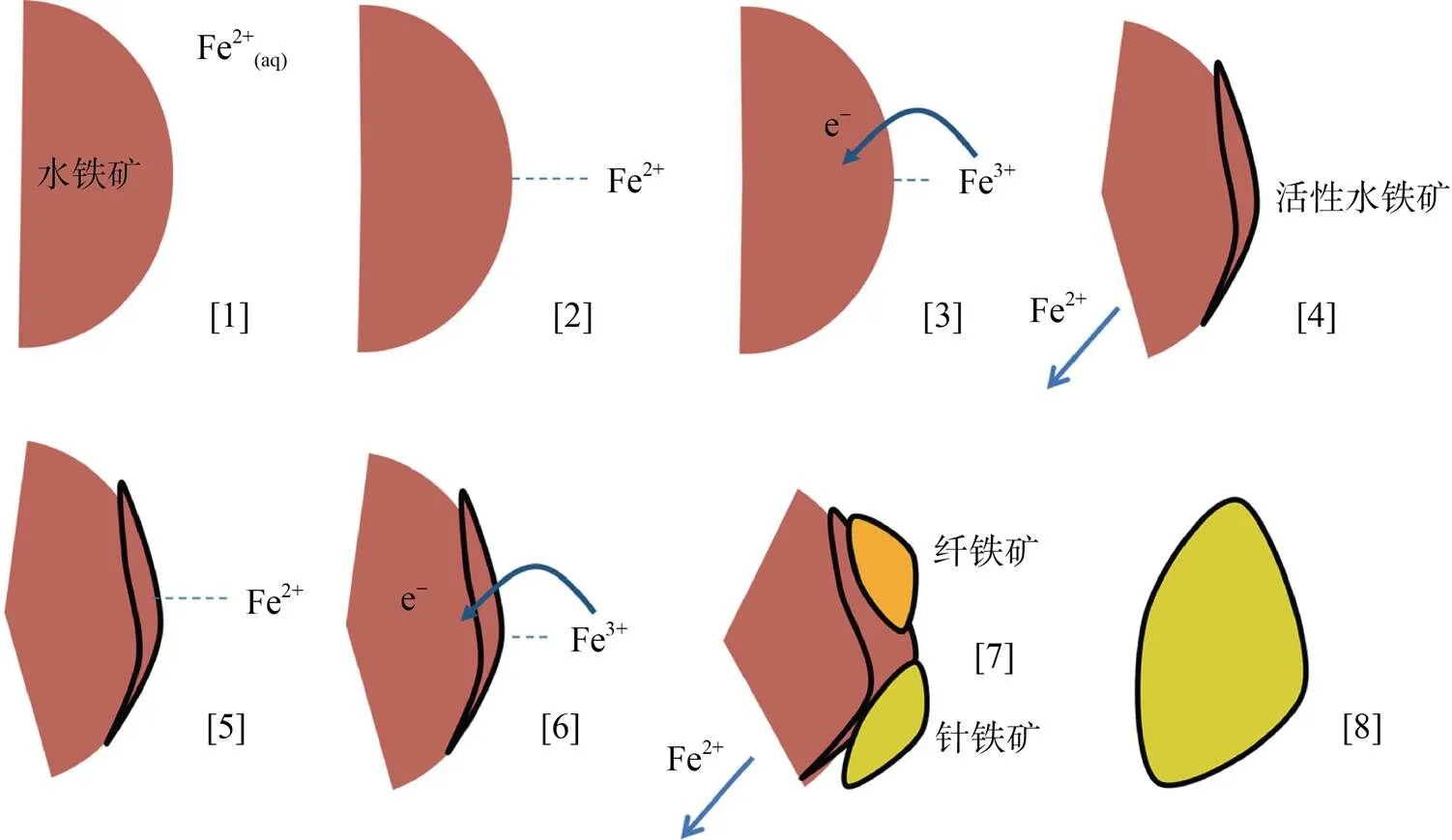
图3 Boland等提出的水铁矿转变机理模型(修改自Boland等[56])
4.2 亚铁催化铁氧化物相变的影响因素
Fe(Ⅱ) 催化铁氧化物相变受水溶液pH及Fe(Ⅱ) 浓度影响。其中,pH通过影响Fe(Ⅱ) 在矿物表面的吸附行为来影响Fe(Ⅱ) 与矿物之间的电子传递过程,从而控制铁氧化物的晶相转化过程。Fe(Ⅱ) 在铁氧化物表面的吸附会产生H+,随着溶液pH的升高,H+浓度降低,越有利于Fe(Ⅱ) 的吸附,从而加快铁氧化物的相变速率。另一方面,溶液pH也会控制铁氧化物相变的矿物类型,Boland等人[56]研究了一定pH范围内(6.17 ~ 7.26),Fe(Ⅱ)催化水铁矿相变反应,发现水铁矿相变的中间产物纤铁矿相通常在较低的 pH条件下普遍存在,较高pH通常会导致相变的最终产物针铁矿的快速形成。而在Hancel等人[57]的研究中,发现pH=8时会促使水铁矿完全转化为磁铁矿沉淀。另一方面,溶液Fe(Ⅱ) 的浓度也能影响铁氧化物相变产物类型。研究表明在低浓度Fe(Ⅱ) (<1.0 mmol/g水铁矿)条件下,水铁矿通过溶解和再沉淀转化为纤铁矿和针铁矿;然而,在较高浓度Fe(Ⅱ) (>1.0 mmol/g水铁矿)条件下,则有利于磁铁矿的沉淀的形成[57-58]。
有机质的存在会抑制Fe(Ⅱ) 催化铁氧化物的相变反应。Jones 等人[55]证明富里酸抑制 Fe(Ⅱ) 催化铁氧化物转化为更稳定的矿物相,反应条件为1 mmol/L Fe(Ⅱ),C/Fe摩尔比约0.6和4.2;类似地,Pasakarnis[59]在1mmol/L Fe(Ⅱ),C/Fe 摩尔比为1和2条件下,发现OM可以防止Fe(Ⅱ) 催化 OM–水铁矿共沉淀物转化为磁铁矿或针铁矿。Chen等人[60]在研究Fe(Ⅱ) 催化有机物–水铁矿共沉淀试验中,发现有机物含量越高对水铁矿相变的抑制作用越明显,当C/Fe 摩尔比大于1.6时,水铁矿表面的吸附位点几乎被有机质占据,此时水铁矿的晶体转化完全被抑制,同时该研究还表明有机物的存在促使水铁矿相变形成纤铁矿。此外,有机质中存在多种官能团,Fe(Ⅱ)可以与这些官能团结合,从而降低了在铁氧化物表面的吸附;另一方面,有机质吸附于铁氧化物表面,能够阻碍铁氧化物的成核作用,从而抑制水铁矿的晶相转变[61]。
金属阳离子的存在也会对 Fe(Ⅱ) 催化铁氧化物相变有着重要影响。在此类研究中,二价金属阳离子备受关注。例如,在一些二价阳离子如 Zn(Ⅱ)、Cu(Ⅱ) 和Mn(Ⅱ) 的存在下,由于离子半径接近 Fe(Ⅱ),导致 Fe(Ⅱ) 催化水铁矿形成了阳离子替代Fe(Ⅱ) 的磁铁矿[62]。Liu等人[63]研究了Mg(Ⅱ)、Ca(Ⅱ)、Ba(Ⅱ)、Mn(Ⅱ)、Co(Ⅱ)、Ni(Ⅱ)和Zn(Ⅱ) 7种金属阳离子对Fe(Ⅱ) 催化水铁矿转化过程的影响,结果发现阳离子与水铁矿之间结合能力显著影响水铁矿的相变过程,具有较高结合能力的阳离子降低了水铁矿上Fe(Ⅱ) 的吸附量,并降低了反应体系的氧化还原电位,从而抑制了水铁矿的转化;此外,这些离子也影响了水铁矿相变的产物类型,在具有较低结合能力的阳离子存在时,水铁矿先转化为纤铁矿,然后转化为针铁矿和磁铁矿,而在具有更高结合能力的阳离子存在下,水铁矿直接转化为针铁矿和磁铁矿。除上述二价金属阳离子,Al3+的作用也不容忽视,铁氧化物发生铝同晶替代是土壤中普遍的矿物学现象。Hansel等人[64]研究了Al替代以及Al吸附对Fe(Ⅱ) 催化水铁矿相变的影响,发现Al替代以及Al吸附都会抑制水铁矿的相变,Al替代或Al吸附量越高,抑制作用越强;同时,Fe(Ⅱ)催化水铁矿相变产生的次生矿物类型也受控于Al替代或Al吸附量,发现Al取代会阻碍纤铁矿形成和磁铁矿成核,而Al 吸附完全抑制针铁矿的形成,对磁铁矿成核的影响较小。
4.3 亚铁催化铁氧化物相变的环境效应
铁氧化物的晶相转化,会导致其表面性质以及反应活性的改变,从而影响与其相关联的重金属和有机质在环境中的地球化学行为。亚铁催化铁氧化物的相变过程中,会导致与铁氧化物共存的重金属离子发生吸附解吸、氧化还原、共沉淀、掺杂等众多反应,进而影响这些重金属离子的迁移转化。Pederen等人[65]研究发现,Fe(Ⅱ) 催化水铁矿、纤铁矿形成的新生铁氧化物对砷酸盐的吸附能力更强,认为缺氧条件下Fe(Ⅱ) 催化铁氧化物重结晶可能是As固定的重要机制。Hu等人[66]发现Fe(Ⅱ) 催化水铁矿相变过程中,溶液中的As掺杂进入新形成的矿物晶格内部。刘承帅等人[15]研究了Pb(Ⅱ) 共存下,Fe(Ⅱ) 催化水铁矿相变过程,发现在水铁矿晶相转变过程中,部分吸附在水铁矿表面的 Pb(Ⅱ) 通过晶体包裹或Fe结构位取代的方式被固定,从而降低了Pb(Ⅱ) 的移动性。Boland等人[67]研究表明Fe(Ⅱ) 催化水铁矿相变过程中,会导致U(Ⅵ) 还原为U(Ⅴ) 并固定于新形成的针铁矿结构中。而另一方面,上述这些被固定的重金属会在铁氧化物发生微生物或化学溶解时释放到土壤环境中,造成相应的重金属环境污染。
铁氧化物很少单独存在,常与环境中大量存在的有机质组成复合物。据估计,沉积物中大约21.5% 的有机碳与Fe通过共沉淀或螯合的形式共存[68],铁氧化物与有机质组成的复合物能够有效抑制微生物对有机物的分解,从而有利于环境中有机碳的固存。Fe(Ⅱ) 催化水铁矿相变反应会间接影响有机质的保存,因为水铁矿具有极大的比表面积,相比相变产生的高结晶度的铁氧化物,水铁矿有利于吸附或共沉淀更多的有机质。对此,Chen等[60]研究认为还原环境下,Fe(Ⅱ) 催化的水铁矿相变过程可能会降低自然环境中的有机质的稳定性。此外,Fe(Ⅱ)催化水铁矿相变也会间接影响微生物对铁氧化物的利用性,通常,结晶度较低的水铁矿更易于被微生物所还原利用。因此,Fe(Ⅱ) 催化水铁矿相变形成结晶态铁氧化物将会抑制微生物对铁的还原利用,进而影响参与微生物铁呼吸的元素的地球化学行为。
5 展望
目前对于铁氧化物–亚铁之间的相互作用的反应机制及其环境影响的室内研究取得了众多好的结果与认识,但由于自然土壤环境条件复杂多变,参与反应的物质种类众多,当前的研究尚有不足,在未来的研究中,以下几个方面值得关注。
1)实际土壤沉积物环境中难以存在纯的铁氧化物,常会伴随离子或分子的掺杂,且铁氧化物常与有机质复合,而目前的研究仅限于实验室合成的纯铁氧化物体系,需进一步模拟研究自然环境中铁氧化物对Fe(Ⅱ) 氧化成矿的影响。另一方面,铁氧化物存在下,Fe(Ⅱ) 氧化形成的新生铁氧化物的性质,包括矿物结构、形貌、粒径等矿物学特征以及生物化学反应活性,还需更加深入的研究。
2)目前关于Fe(Ⅱ) –铁氧化物还原污染物以及Fe(Ⅱ) 催化铁氧化物相变耦合重金属迁移转化的认识尚限于较为单一的实验条件,如何将实验所取得的认识应用于复杂动态的自然系统中,仍需扩展实验反应参数,并基于热力学和动力学数据,建立反应模型,模拟实际环境反应发生,预测污染物在土壤中的行为。
3)一些土壤环境存在重金属与有机污染物组成的复合污染,目前关于研究Fe(Ⅱ)–铁氧化物系统在复合污染物系统中的表现尚显不足,需要进一步研究关注,以探索Fe(Ⅱ)–铁氧化物系统在处理该类污染的可行性。
4)当前的研究大多以室内实验室模拟反应为主,未来的研究需关注自然土壤中,铁氧化物与溶液相亚铁的界面反应及环境影响。鉴于实际土壤系统的复杂性,目前常规分析表征技术在应用于实地土壤界面反应微观尺度的研究仍有众多困难,需要开发更加适用于实际土壤环境的原位观测技术手段和方法。
[1] Kappler A, Schink B, Newman D K. Fe(Ⅲ) mineral formation and cell encrustation by the nitrate-dependent Fe(Ⅱ)-oxidizer strain BoFeN1[J]. Geobiology, 2005, 3(4): 235–245.
[2] 胡敏, 李芳柏. 土壤微生物铁循环及其环境意义[J]. 土壤学报, 2014, 51(4): 683–698.
[3] Buerge I J, Hug S J. Influence of mineral surfaces on chromium(Ⅵ) reduction by iron(Ⅱ)[J]. Environmental Science & Technology, 1999, 33(23): 4285–4291.
[4] Charlet L, Bosbach D, Peretyashko T. Natural attenuation of TCE, As, Hg linked to the heterogeneous oxidation of Fe(Ⅱ): An AFM study[J]. Chemical Geology, 2002, 190(1/2/3/4): 303–319.
[5] Amonette J E, Workman D J, Kennedy D W, et al. Dechlorination of carbon tetrachloride by Fe(Ⅱ) associated with goethite[J]. Environmental Science & Technology, 2000, 34(21): 4606–4613.
[6] Chun C L, Penn R L, Arnold W A. Kinetic and microscopic studies of reductive transformations of organic contaminants on goethite[J]. Environmental Science & Technology, 2006, 40(10): 3299–3304.
[7] Elsner M, Schwarzenbach R P, Haderlein S B. Reactivity of Fe(Ⅱ)-bearing minerals toward reductive transformation of organic contaminants[J]. Environmental Science & Technology, 2004, 38(3): 799–807.
[8] Jones A M, Kinsela A S, Collins R N, et al. The reduction of 4-chloronitrobenzene by Fe(Ⅱ)-Fe(Ⅲ) oxide systems - correlations with reduction potential and inhibition by silicate[J]. Journal of Hazardous Materials, 2016, 320: 143–149.
[9] Pecher K, Haderlein S B, Schwarzenbach R P. Reduction of polyhalogenated methanes by surface-bound Fe(Ⅱ) in aqueous suspensions of iron oxides[J]. Environmental Science & Technology, 2002, 36(8): 1734–1741.
[10] Tai Y L, Dempsey B A. Nitrite reduction with hydrous ferric oxide and Fe(Ⅱ): Stoichiometry, rate, and mechanism[J]. Water Research, 2009, 43(2): 546–552.
[11] Larese-Casanova P, Kappler A, Haderlein S B. Heterogeneous oxidation of Fe(Ⅱ) on iron oxides in aqueous systems: Identification and controls of Fe(Ⅲ) product formation[J]. Geochimica et Cosmochimica Acta, 2012, 91: 171–186.
[12] Williams A G B, Scherer M M. Spectroscopic evidence for Fe(Ⅱ)–Fe(Ⅲ) electron transfer at the iron oxide–water interface[J]. Environmental Science & Technology, 2004, 38(18): 4782–4790.
[13] Handler R M, Beard B L, Johnson C M, et al. Atom exchange between aqueous Fe(Ⅱ) and goethite: An Fe isotope tracer study[J]. Environmental Science & Technology, 2009, 43(4): 1102–1107.
[14] Yanina S V, Rosso K M. Linked reactivity at mineral-water interfaces through bulk crystal conduction[J]. Science, 2008, 320(5873): 218–222.
[15] 刘承帅, 李芳柏, 陈曼佳, 等. Fe(Ⅱ)催化水铁矿晶相转变过程中Pb的吸附与固定[J]. 化学学报, 2017, 75(6): 621–628.
[16] Tartaj P, Morales M P, Gonzalez-Carreño T, et al. The iron oxides strike back: From biomedical applications to energy storage devices and photoelectrochemical water splitting[J]. Advanced Materials, 2011, 23(44): 5243–5249.
[17] Cornell R M, Schwertmann U. The iron oxides: Structure, properties, reactions, occurrences, and uses[M]. Weinheim: Wiley-VCH, 2003.
[18] Hiemstra T. Formation, stability, and solubility of metal oxide nanoparticles: Surface entropy, enthalpy, and free energy of ferrihydrite[J]. Geochimica et Cosmochimica Acta, 2015, 158: 179–198.
[19] Liao S, Wang X M, Yin H, et al. Effects of Al substitution on local structure and morphology of lepidocrocite and its phosphate adsorption kinetics[J]. Geochimica et Cosmochimica Acta, 2020, 276: 109–121.
[20] Chorover J, Amistadi M K. Reaction of forest floor organic matter at goethite, birnessite and smectite surfaces[J]. Geochimica et Cosmochimica Acta, 2001, 65(1): 95–109.
[21] Huang X P, Hou X J, Zhang X, et al. Facet-dependent contaminant removal properties of hematite nanocrystals and their environmental implications[J]. Environmental Science: Nano, 2018, 5(8): 1790–1806.
[22] Navrotsky A, Mazeina L, Majzlan J. Size-driven structural and thermodynamic complexity in iron oxides[J]. Science, 2008, 319(5870): 1635–1638.
[23] 胡世文. 铁矿物生物和非生物转化过程中砷和有机碳的微观固存机制[D]. 广州: 华南理工大学, 2021.
[24] Huang J Z, Jones A, Waite T D, et al. Fe(Ⅱ) redox chemistry in the environment[J]. Chemical Reviews, 2021, 121(13): 8161–8233.
[25] Melton E D, Swanner E D, Behrens S, et al. The interplay of microbially mediated and abiotic reactions in the biogeochemical Fe cycle[J]. Nature Reviews Microbiology, 2014, 12(12): 797–808.
[26] Morgan B, Lahav O. The effect of pH on the kinetics of spontaneous Fe(Ⅱ) oxidation by O2in aqueous solution - basic principles and a simple heuristic description[J]. Chemosphere, 2007, 68(11): 2080–2084.
[27] Park U, Dempsey B A. Heterogeneous oxidation of Fe(Ⅱ) on ferric oxide at neutral pH and a low partial pressure of O2[J]. Environmental Science & Technology, 2005, 39(17): 6494–6500.
[28] Tamura H, Kawamura S, Hagayama M. Acceleration of the oxidation of Fe2+ions by Fe(Ⅲ)-oxyhydroxides[J]. Corrosion Science, 1980, 20(8/9): 963–971.
[29] Jones A M, Griffin P J, Collins R N, et al. Ferrous iron oxidation under acidic conditions-The effect of ferric oxide surfaces[J]. Geochimica et Cosmochimica Acta, 2014, 145: 1–12.
[30] Chen C M, Thompson A. The influence of native soil organic matter and minerals on ferrous iron oxidation[J]. Geochimica et Cosmochimica Acta, 2021, 292: 254–270.
[31] Chen C M, Thompson A. Ferrous iron oxidation under varying pO2levels: The effect of Fe(Ⅲ)/Al(Ⅲ) oxide minerals and organic matter[J]. Environmental Science & Technology, 2018, 52(2): 597–606.
[32] Zwank L, Elsner M, Aeberhard A, et al. Carbon isotope fractionation in the reductive dehalogenation of carbon tetrachloride at iron (hydr)oxide and iron sulfide minerals[J]. Environmental Science & Technology, 2005, 39(15): 5634–5641.
[33] Maithreepala R A, Doong R A. Synergistic effect of copper ion on the reductive dechlorination of carbon tetrachloride by surface-bound Fe(Ⅱ) associated with goethite[J]. Environmental Science & Technology, 2004, 38(1): 260–268.
[34] Colón D, Weber E J, Anderson J L. Effect of natural organic matter on the reduction of nitroaromatics by Fe(Ⅱ) species[J]. Environmental Science & Technology, 2008, 42(17): 6538–6543.
[35] Luan F B, Xie L, Li J, et al. Abiotic reduction of nitroaromatic compounds by Fe(Ⅱ) associated with iron oxides and humic acid[J]. Chemosphere, 2013, 91(7): 1035–1041.
[36] Jeon B H, Dempsey B A, Burgos W D, et al. Chemical reduction of U(Ⅵ) by Fe(Ⅱ) at the solid-water interface using natural and synthetic Fe(Ⅲ) oxides[J]. Environmental Science & Technology, 2005, 39(15): 5642–5649.
[37] Peretyazhko T, Zachara J M, Heald S M, et al. Heterogeneous reduction of Tc(Ⅶ) by Fe(Ⅱ) at the solid-water interface[J]. Geochimica et Cosmochimica Acta, 2008, 72(6): 1521–1539.
[38] Jang J H, Dempsey B A, Burgos W D. Reduction of U(Ⅵ) by Fe(Ⅱ) in the presence of hydrous ferric oxide and hematite: Effects of solid transformation, surface coverage, and humic acid[J]. Water Research, 2008, 42(8/9): 2269–2277.
[39] Hering J, Stumm W. Oxidative and reductive dissolution of minerals[J]. Reviews in Mineralogy & Geochemistry, 1990, 23: 427–465.
[40] Orsetti S, Laskov C, Haderlein S B. Electron transfer between iron minerals and quinones: Estimating the reduction potential of the Fe(Ⅱ)-goethite surface from AQDS speciation[J]. Environmental Science & Technology, 2013, 47(24): 14161–14168.
[41] Silvester E, Charlet L, Tournassat C, et al. Redox potential measurements and Mössbauer spectrometry of FeⅡadsorbed onto FeⅢ(oxyhydr)oxides[J]. Geochimica et Cosmochimica Acta, 2005, 69(20): 4801–4815.
[42] Gorski C A, Scherer M M. Influence of magnetite stoichiometry on Fe(Ⅱ) uptake and nitrobenzene reduction[J]. Environmental Science & Technology, 2009, 43(10): 3675–3680.
[43] Larese-Casanova P, Scherer M M. Fe(Ⅱ) sorption on hematite: New insights based on spectroscopic measurements[J]. Environmental Science & Technology, 2007, 41(2): 471–477.
[44] Rosso K M, Yanina S V, Gorski C A, et al. Connecting observations of hematite (α-Fe2O3) growth catalyzed by Fe(Ⅱ)[J]. Environmental Science & Technology, 2010, 44(1): 61–67.
[45] Felmy A R, Ilton E S, Rosso K M, et al. Interfacial reactivity of radionuclides: Emerging paradigms from molecular-level observations[J]. Mineralogical Magazine, 2011, 75(4): 2379–2391.
[46] Felmy A R, Moore D A, Rosso K M, et al. Heterogeneous reduction of PuO2with Fe(Ⅱ): Importance of the Fe(Ⅲ) reaction product[J]. Environmental Science & Technology, 2011, 45(9): 3952–3958.
[47] Stemig A M, Do T A, Yuwono V M, et al. Goethite nanoparticle aggregation: Effects of buffers, metal ions, and 4-chloronitrobenzene reduction[J]. Environmental Science: Nano, 2014, 1(5): 478–487.
[48] Cwiertny D M, Handler R M, Schaefer M V, et al. Interpreting nanoscale size-effects in aggregated Fe-oxide suspensions: Reaction of Fe(Ⅱ) with goethite[J]. Geochimica et Cosmochimica Acta, 2008, 72(5): 1365–1380.
[49] Zhang Y, Charlet L, Schindler P W. Adsorption of protons, Fe(Ⅱ) and Al(Ⅲ) on lepidocrocite (γ-FeOOH)[J]. Colloids and Surfaces, 1992, 63(3/4): 259–268.
[50] Liger E, Charlet L, Van Cappellen P. Surface catalysis of uranium(Ⅵ) reduction by iron(Ⅱ)[J]. Geochimica et Cosmochimica Acta, 1999, 63(19/20): 2939–2955.
[51] Charlet L, Silvester E, Liger E. N-compound reduction and actinide immobilisation in surficial fluids by Fe(Ⅱ): The surface FeⅢOFeⅡOH° species, as major reductant[J]. Chemical Geology, 1998, 151(1/2/3/4): 85–93.
[52] Zhang H C, Taujale S, Huang J Z, et al. Effects of NOM on oxidative reactivity of manganese dioxide in binary oxide mixtures with goethite or hematite[J]. Langmuir: the ACS Journal of Surfaces and Colloids, 2015, 31(9): 2790–2799.
[53] Vindedahl A M, Stemig M S, Arnold W A, et al. Character of humic substances as a predictor for goethite nanoparticle reactivity and aggregation[J]. Environmental Science & Technology, 2016, 50(3): 1200–1208.
[54] 胡世文, 刘同旭, 李芳柏, 等. 土壤铁矿物的生物-非生物转化过程及其界面重金属反应机制的研究进展[J]. 土壤学报, 2022, 59(1): 54–65.
[55] Jones A M, Collins R N, Rose J, et al. The effect of silica and natural organic matter on the Fe(Ⅱ)-catalysed transformation and reactivity of Fe(Ⅲ) minerals[J]. Geochimica et Cosmochimica Acta, 2009, 73(15): 4409–4422.
[56] Boland D D, Collins R N, Miller C J, et al. Effect of solution and solid-phase conditions on the Fe(Ⅱ)-accelerated transformation of ferrihydrite to lepidocrocite and goethite[J]. Environmental Science & Technology, 2014, 48(10): 5477–5485.
[57] Hansel C M, Benner S G, Fendorf S. Competing Fe (Ⅱ)-induced mineralization pathways of ferrihydrite[J]. Environmental Science & Technology, 2005, 39(18): 7147–7153.
[58] Hansel C M, Benner S G, Neiss J, et al. Secondary mineralization pathways induced by dissimilatory iron reduction of ferrihydrite under advective flow[J]. Geochimica et Cosmochimica Acta, 2003, 67(16): 2977–2992.
[59] Pasakarnis T S. Effects of carbon during Fe(Ⅱ)-catalyzed Fe oxide recrystallization[D]. The University of Iowa, Doctor of Philosophy, 2013. DOI: 10.17077/etd.wnhgjk73
[60] Chen C M, Kukkadapu R, Sparks D L. Influence of coprecipitated organic matter on Fe2+(aq)-catalyzed transformation of ferrihydrite: Implications for carbon dynamics[J]. Environmental Science & Technology, 2015, 49(18): 10927–10936.
[61] Xiao W, Jones A M, Collins R N, et al. Investigating the effect of ascorbate on the Fe(Ⅱ)-catalyzed transformation of the poorly crystalline iron mineral ferrihydrite[J]. Biochimica et Biophysica Acta (BBA) - General Subjects, 2018, 1862(8): 1760–1769.
[62] Jang J H, Dempsey B A, Catchen G L, et al. Effects of Zn(Ⅱ), Cu(Ⅱ), Mn(Ⅱ), Fe(Ⅱ), NO3−, or SO42−at pH 6.5 and 8.5 on transformations of hydrous ferric oxide (HFO) as evidenced by Mössbauer spectroscopy[J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2003, 221(1/2/3): 55–68.
[63] Liu C S, Zhu Z K, Li F B, et al. Fe(Ⅱ)-induced phase transformation of ferrihydrite: The inhibition effects and stabilization of divalent metal cations[J]. Chemical Geology, 2016, 444: 110–119.
[64] Hansel C M, Learman D R, Lentini C J, et al. Effect of adsorbed and substituted Al on Fe(Ⅱ)-induced mineralization pathways of ferrihydrite[J]. Geochimica et Cosmochimica Acta, 2011, 75(16): 4653–4666.
[65] Pedersen H D, Postma D, Jakobsen R. Release of arsenic associated with the reduction and transformation of iron oxides[J]. Geochimica et Cosmochimica Acta, 2006, 70(16): 4116–4129.
[66] Hu S W, Lu Y, Peng L F, et al. Coupled kinetics of ferrihydrite transformation and As(Ⅴ) sequestration under the effect of humic acids: A mechanistic and quantitative study[J]. Environmental Science & Technology, 2018, 52(20): 11632–11641.
[67] Boland D D, Collins R N, Glover C J, et al. Reduction of U(Ⅵ) by Fe(Ⅱ) during the Fe(Ⅱ)-accelerated transformation of ferrihydrite[J]. Environmental Science & Technology, 2014, 48(16): 9086–9093.
[68] Lalonde K, Mucci A, Ouellet A, et al. Preservation of organic matter in sediments promoted by iron[J]. Nature, 2012, 483(7388): 198–200.
Soil Iron Oxide-Ferrous Interaction and Its Environmental Effects: A Review
YAO Yuan1,2, YU Guanghui1, TENG Hui1*
(1Institute of Surface-Earth System Science, School of Earth System Science, Tianjin University, Tianjin 300072, China; 2 Xi’an Center of China Geological Survey, Xi’an 710119, China)
Iron (hydr)oxides (hereafter termed iron oxides) catalyzed oxidation of aqueous Fe(Ⅱ) is an important process in soil environments not only because the two phases often coexist but also because the reactions are frequently coupled with the reduction of organic and inorganic contaminants. Concurrently, recrystallization of iron oxides in the presence of aqueous Fe(Ⅱ) plays a critical part in the iron geochemical cycling as the process leads to changes in the structure and surface properties of iron oxides to alter the ensuing adsorption of heavy metals and organic matter in soils and sediments. In this paper, we reviewed the recent progresses in understanding the interactions of iron oxides and aqueous ferrous with a focus on the reaction mechanism and controlling factors for: 1) iron oxide-catalyzed Fe(Ⅱ) oxidation; 2) contaminants reduction by the aqueous Fe(Ⅱ)-iron oxides system; and 3) Fe(Ⅱ)-catalyzed recrystallization of iron oxides. In addition, we provide a perspective for the future research of the iron oxide-aqueous Fe(Ⅱ) interface reaction and related environmental impact in soils.
Iron oxide; Fe(Ⅱ) oxidation; Mineral transformation; Organic contaminants; Heavy metals
X142
A
10.13758/j.cnki.tr.2023.04.004
姚远, 余光辉, 滕辉. 土壤铁氧化物–亚铁的相互作用及其环境影响研究进展. 土壤, 2023, 55(4): 718–728.
国家自然科学基金重点项目(41830859)资助。
(huihenry.teng@tju.edu.cn)
姚远(1997—),男,陕西西安人,硕士,主要研究方向为土壤铁矿物的形成及其环境效应。E-mail: YaoYuanSF@163.com
