帕金森病发病机制研究进展
2023-09-22肖琪樊慧杰李艳荣孙芮芮贾璐徐磊尉杰忠肖保国马存根柴智
肖琪,樊慧杰,李艳荣,孙芮芮,贾璐,徐磊,尉杰忠,肖保国,马存根,柴智*
1山西中医药大学基础医学院/神经生物学研究中心,山西晋中 030619;2山西大同大学脑科学研究所,山西大同037009;3复旦大学华山医院神经病学研究所,上海 200025
帕金森病(Parkinson's disease,PD)也称震颤麻痹(paralysis agitans,shaking palsy),是中老年人常见的神经系统慢性变性疾病,也是常见的锥体外系疾病。PD 的病理标志是α-突触核蛋白(α-synuclein,α-syn)聚集,尤其在黑质内;这一聚集以细胞内的路易体和神经炎聚集的形式发生[1]。PD 患者脑中的显著病理特征为多巴胺(dopamine,DA)能神经元慢性丢失或黑质内尚存的DA 能神经元中的异常聚集体[2]。65 岁以上人群PD 患病率达1000/10 万,且随年龄增长而增高,男性稍多于女性。该病的主要临床表现包括静止性震颤,动作迟缓及减少,肌张力增高,姿势不稳等[3-4]。PD的发病原因十分复杂,本文就导致PD的致病机制及其治疗进展做一综述,以期为相关研究提供参考。
1 α-syn与PD
α-syn 的病理变化是PD 的标志,也会进一步导致神经元功能障碍和死亡。α-syn水平改变可导致家族性PD,其积累可导致突触核蛋白病,包括PD、路易体痴呆和多系统萎缩[5]。
1.1 α-syn异常聚集 α-syn是由140个氨基酸残基组成的细胞内神经元蛋白,主要位于突触前末端;该突触末端可介导神经递质的释放,也可导致神经元损伤,并参与中枢神经系统的神经变性过程,是路易体和路易突起包含的主要成分[6]。在单体状态下,α-syn 是无序、可溶的,是细胞质中的常见形式[7]。α-syn低聚物是路易体的淀粉样蛋白生成途径中的主要毒性物质[8]。在体的一种新型肽模拟物FN075 可促进内源性α-syn 聚集,注射FN075 到大鼠黑质后,会导致其运动功能障碍及DA能神经元丢失,提示αsyn 聚集与PD 的发生有关[9]。敲低小胶质细胞中一种氧化应激传感器(DJ-1)后,其对DA 能神经元的神经毒性增加,且对α-syn产生炎性反应,使细胞摄取和清除α-syn的能力受损[10]。Hua等[11]研究发现,星形胶质细胞通过内吞作用捕获α-syn,并将其运输到溶酶体发挥清除作用,若捕获能力长期大于降解能力,可使α-syn 在星形胶质细胞内聚集,导致PD 的发生。病理性α-syn错误折叠形成的聚集体可在淋巴细胞活化基因3(lymphocyte activation gene 3,LAG3)的作用下,在相邻神经元细胞之间传播,加速神经元细胞的损伤,导致PD的发生与进行性发展[12]。以上均提示α-syn 的异常聚集是导致PD 的重要机制(图1),因此,如何阻止α-syn 异常聚集成为相关研究关注的核心问题之一。
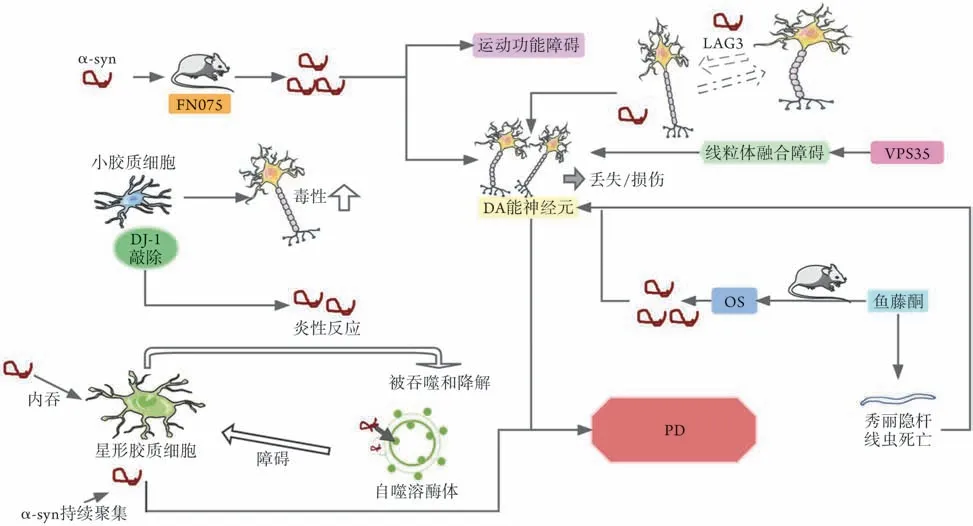
图1 α-突触核蛋白异常聚集导致帕金森病的可能机制Fig.1 The pathogenesis of Parkinson's disease caused by anomalous aggregation of α-synuclein
针对α-syn 的异常聚集,目前提出了抑制α-syn聚集和促进α-syn消除等措施。例如,蛋白酶体相关的去泛素化酶Usp14 的小分子抑制剂可通过增强蛋白酶体活性促进α-syn的清除[13];姜黄素可提升细胞自噬能力,使α-syn 出现自噬性清除,从而减轻DA能神经元损伤[14]。近期阻止α-syn聚集的相关研究不断增多,有望推动PD治疗方式的进步。
1.2SNCA基因(α-syn基因)点突变SNCA位于人类染色体4q21-23,包含6 个外显子,该基因编码蛋白质α-syn,是发现较早的与家族性PD 相关的致病基因。研究显示,SNCA的点突变可引发α-syn 异常折叠、堆积及神经元损伤,继而导致PD的发生[15]。
1.2.1 A53T点突变 1997年,有研究报告常染色体显性遗传PD家系SNCAA53T突变后,人们开始对不同PD 人群进行SNCA基因突变检测,并制作了多种SNCA突变的动物模型,如Thy1-α SNCAA53T转基因小鼠模型、Thy1-α SNCAWT转基因小鼠模型、α-syn转基因大鼠PD 模型(Thy-A30P-A53T)、α-syn 过表达大鼠PD模型(rAAV-CBA-α-synuclein)等。这些模型能模仿PD 的主要生理病理特征和功能障碍,提示SNCA突变与α-syn改变及神经元变性有关(表1)。徐美玲等[16]的研究显示,A53T转基因小鼠发生了胃肠道局部和系统性的炎症反应及氧化应激反应,存在α-syn 的异常聚集物和修饰形式,在黑质和纹状体中,α-syn蛋白表达升高,提示α-syn的A53T点突变参与了PD 的发病过程。吴婷等[17]报告,SNCA基因A53T 点位突变会引发小鼠中脑DA 能神经元的DNA甲基化改变,而在PD 患者中,SNCA基因甲基化水平下降,导致其表达产物α-syn增多。而通过慢病毒转染建立的α-syn A53T过表达SHSY5Y细胞模型可模仿PD神经元α-syn过表达这一病理特征[18]。A53T突变的部分患者具有更多的突触核蛋白沉积,与野生型相比,α-syn A53T 突变对原纤维形成蛋白质起到明显的加速作用。研究显示,在甲基-苯基-吡啶离子(1-methyl-4-phenylpyr-idinium,MPP+)诱导的PD SHSY5Y 细胞模型中,银杏内酯B(ginkgolide B,GB)可下调 miR-207的靶基因LAMP2A,通过分子伴侣介导的自噬作用促进α-syn 降解,改善野生型 α-syn 聚集体导致的神经细胞凋亡[19-20]。总之,SNCAA53T突变研究为PD发病机制与治疗的探索提供了一个新方向。

表1 与帕金森病相关的SNCA基因点突变Tab.1 SNCA point mutations related to Parkinson's disease
1.2.2 A30P点突变 α-A30P转基因大鼠模型中,大鼠中脑黑质致密部DA 能神经元发生了大量变性丢失,且α-syn在神经元缺失区域内大量聚集,提示该模型可模拟PD 疾病的主要病理特点,也可模拟PD患者的部分运动功能障碍[21](表1)。已知PD 的发生发展与大脑海马齿状回(dentate gyrus,DG)神经紊乱有关,而研究显示A30PSNCA可显著抑制小鼠DG细胞的增殖、分化等神经发生活动[22]。人α-A30P突变型转基因大鼠PD 模型脑内出现大量聚集的α-syn 蛋白,DA能神经元数量减少,进而导致进行性的运动功能障碍[23]。
1.2.3 G51D 点突变 G51D 点突变于2013 年由苏珊娜·勒萨热等发现。SNCAG51D 点突变在中国汉族人群中十分罕见,且在全球范围也较为少见。国内解放军总医院海南医院报告了2 例SNCAG51D 错义突变的PD 患者,其细胞病理具有PD 和多系统萎缩(multiple system atrophy,MSA)的特征,有明显的皮质萎缩。临床研究显示,G51D 病例存在α-syn 异常聚集,具有早发性左旋多巴反应性PD 综合征伴痴呆、幻觉和自主神经功能障碍[24]。在国外4例G51D突变患者的临床调查中,其中3例早发性PD患者均伴有运动障碍和锥体束受累的表现,病理检查结果显示黑质和纹状体中存在大量神经元丢失,并伴有星形胶质细胞增生[25](表1)。SNCAG51D突变病例始终具有PD 和MSA 的临床和神经病理学特征,虽然H50Q 突变位点紧邻α-syn 区域内的G51D 位点,但SNCAG51D 突变的PD 患者与携带H50Q 的PD 患 者存在明显区别。
1.2.4 H50Q点突变 2013年,H50Q点突变作为PD的新型致病性突变被提出。有研究指出,H50Q突变可加速体外α-syn 原纤维的形成、细胞外α-syn 的神经元毒性并导致线粒体功能障碍[26-27](表1)。SNCAH50Q突变病例的临床表现与迟发性特发性PD相似,表现出持续左旋多巴反应。
2 线粒体功能障碍与PD
线粒体功能障碍会损伤神经元和神经胶质细胞,导致腺苷三磷酸(ATP)减少、自由基增多、钙稳态失衡、线粒体DNA(mitochondrial DNA,mtDNA)突变,以及通过多种途径与线粒体对接的异常蛋白质产生,进而引起PD的神经变性。
2.1 线粒体动力学受损 线粒体融合与裂变动力学功能失调被认为是导致神经退行性疾病(如PD)的原因之一。裂变可促进线粒体的分布以响应其对ATP的需求,而融合有助于替换受损的mtDNA。与线粒体功能障碍相关的新的PD基因——液泡分选蛋白35基因(VPS35)缺陷或突变会导致中脑的线粒体融合功能障碍,使线粒体断裂、分解,DA 能神经元损伤,最终引起神经变性[28-29]。线粒体动力学受损会导致活性氧 (reactive oxygen species,ROS) 产生,线粒体膜电位降低,并可能加剧功能失调的线粒体的积累。多项研究显示,线粒体功能障碍是PD发病机制的核心,富含亮氨酸的重复激酶2 基因(leucine-rich repeat kinase 2 gene,LRRK2)突变可影响线粒体动力学、线粒体的运输及粒体自噬。在散发性PD 细胞模型中,调节线粒体裂变事件的蛋白质(Drp1)依赖性线粒体片段化在介导散发性PD 中线粒体DNA 诱导的线粒体异常和细胞功能障碍中起着至关重要的作用[30]。
2.2 线粒体呼吸链功能障碍 线粒体呼吸链抑制可能导致氧消耗不完全、ATP减少和自由基形成增加,自由基则直接抑制线粒体的呼吸链导致有害循环,从而引起细胞氧化损伤。过氧化物酶体增殖物激活受体-γ共激活因子-1α(PGC-1α)是多种转录因子的辅激活因子,它是线粒体生物发生的主要调节剂。在细胞模型中,PGC-1α可上调线粒体呼吸链的核编码亚基,并阻止鱼藤酮或突变体α-syn 诱导的DA 能神经元丢失和神经毒性[31]。研究显示,PD患者死亡后脑中PGC-1α 水平在黑质中降低;另有研究发现,PGC-1α 过表达会导致大鼠黑质纹状体系统中DA 能神经元突然退化[32]。这些都提示线粒体功能障碍对PD疾病的进展有重要作用。在过去几年中,1-甲基-4苯基-1,2,3,6-四氢吡啶(MPTP)和鱼藤酮等诱导线粒体呼吸链功能障碍的模型常被应用于PD的相关研究,以复制PD 的神经病理学特征[33]。有研究发现,鱼藤酮可通过引起转基因线虫zc Is9;ot Is181线粒体损伤,导致线粒体稳态失衡,进而诱发线虫异常死亡及DA 能神经元退化,而Von Hippel- Lindau(VHL)综合征的抑制剂VH298 能减少鱼藤酮导致的PD 线虫异常死亡,可通过保护DA 能神经元而改善由DA能神经元丢失所致的运动和觅食行为异常,为VHL抑制剂治疗PD提供了一定的理论基础[34](图1)。以上多项研究提示,线粒体呼吸链功能障碍与PD的发病密不可分。
2.3 基因突变引起线粒体功能异常 已有研究显示,约有20种PD易感基因(如PRKN、SNCA等)可参与线粒体自噬和线粒体功能的调控过程,这些基因突变导致的遗传性PD患者会出现线粒体自噬通路损伤和线粒体功能障碍。
2.3.1PRKN基因PRKN基因大小为1.3Mb,定位于6 号染色体上,其编码的蛋白质Parkin 包含465 个氨基酸残基。1988年的研究显示,PRKN基因突变会增加散发性、早发性PD 的发生风险。PD 相关的PRKN基因突变可影响线粒体自噬和mtDNA,导致线粒体自噬受损。PRKN突变使受损线粒体增多,黑质中DA 能神经元丧失,最终导致PD 的发生(表2)。研究发现,PRKN 缺乏会造成线粒体自噬受损,导致mtDNA释放,从而引发炎症;白细胞介素-6(IL-6)是一种促炎细胞因子,线粒体自噬受损和随后的mtDNA 释放导致IL-6 水平升高,与PRKN基因突变相关PD的发病机制有关。Borsche等[35]发现,PRKN/PINK1双等位基因突变相关PD的临床特点为发病年龄更早、病程更长、嗅觉障碍减少。
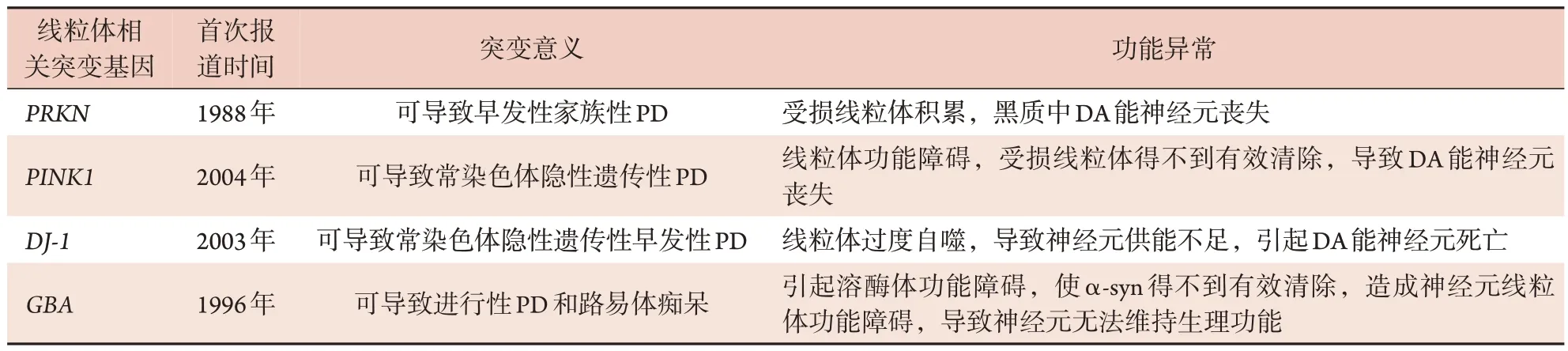
表2 引发与帕金森病相关线粒体功能异常的部分基因突变Tab.2 Gene mutations which lead to mitochondrial dysfunction related of Parkinson's disease
2.3.2PINK1基因PINK1基因定位于1 号染色体,其突变可导致常染色体隐性遗传性PD。2004年的研究显示,PINK1基因突变可消除PINK1基因对细胞的保护作用,导致细胞应激增加。线粒体蛋白PINK1突变的相关研究提示了线粒体功能障碍与PD发病机制的联系。PINK1基因突变可导致细胞线粒体呼吸水平降低、ATP合成减少及PD细胞培养模型的α-syn增高。研究发现,线粒体膜电位丧失可造成PINK1在膜上积累并磷酸化Parkin蛋白,进而导致线粒体自噬。PINK1在线粒体自噬方面表现尤为突出,当PINK1发生突变时,会破坏线粒体生物学的多个部分,从而使线粒体发生功能障碍,导致受损线粒体得不到有效清除,进而引发DA 能神经元的死亡,最终造成PD的发生[36](表2)。
2.3.3DJ-1基因 2003 年,DJ-1基因(又称PARK7基因)被发现是常染色体隐性遗传性早发性PD 综合征(AREP)的致病基因之一。DJ-1 蛋白含189 个氨基酸残基,相对分子质量约为20×103。DJ-1 蛋白的功能包括转录调节、抗氧化反应以及分子伴侣和线粒体调节[37]。DJ-1 能对氧化应激作出反应并防御线粒体损伤。研究显示,DJ-1突变可影响线粒体合成ATP及自噬的过程,且其突变发生在DA 能神经元细胞时,可造成线粒体的过度自噬,导致神经元供能不足,引发DA能神经元死亡(表2)。
2.3.4GBA基因GBA基因位于1 号染色体短臂端,编码溶酶体的β-葡萄糖脑苷脂酶(GCase)。1996 年,GBA基因突变患者被发现具有进行性PD的特征。研究显示,GBA基因突变引起的GCase 表达量及酶活性降低造成溶酶体功能障碍,是导致PD的主要原因之一;其主要影响自噬和伴侣介导自噬过程,导致聚集的α-syn无法清除,最终影响神经元的正常生理活动。GBA突变不仅使α-syn得不到有效清除,还会造成神经元线粒体功能障碍,导致神经元无法维持正常生理功能[38]。
3 氧化应激与PD
氧化应激(oxidative stress,OS)是受到较多关注的一种PD发病机制。通常情况下,ROS的产生和消除是协调一致的,以维持氧化还原状态,一旦该平衡被打破,就会诱发OS,继而可能发生PD 等疾病[39-40]。
3.1 氧化应激损伤 OS 由过量的ROS 引起,这是促氧化和抗氧化稳态失衡的结果[41]。鱼藤酮能通过氧化应激途径使小鼠脑黑质中α-syn聚集,从而导致DA 能神经元损伤。尸检结果显示,PD 患者黑质部位存在严重的OS,如游离铁离子增多,线粒体复合体Ⅰ功能受损,大量被氧化损伤的脂质、蛋白质和DNA,使之出现PD 综合征的表现。这些都提示OS与PD的发生及进展密切相关。超氧化物、过氧化氢(H2O2)和高活性羟基自由基是哺乳动物细胞中主要的ROS,可破坏包括核酸、脂质和蛋白质在内的大分子,导致DA 能神经元变性和神经网络功能障碍,最终发展为PD[42]。因此,抑制正常氧化还原机制所导致的OS 与神经元变性在PD 研究中非常重要。目前通过抗氧化治疗PD的研究越来越多,盐酸多奈哌齐片联合司来吉兰片可抑制OS,降低ROS自由基的生成,阻止DA能神经元的变性坏死[43];雷沙吉兰可防止自由基损伤,对运动缺陷有较好的效果[44];姜黄素可促进沉默信息调节因子3 的表达,清除小胶质细胞源性ROS,从而保护DA 能神经元[45]。此外,关于具有抗氧化作用的中药材及天然抗氧化剂的研究也十分丰富,如茶多酚可抗氧化,抑制α-syn 聚集,调节蛋白激酶信号通路,保护脑组织[46];美多芭也具有良好的抗氧化作用,可清除自由基,缓解PD 患者的症状[47];樟芝多糖可提高抗氧化酶的含量,明显改善PD小鼠的认知和运动行为[48];枸杞多糖能有效减轻PD 小鼠中脑OS 损伤,对黑质致密部DA能神经元起保护作用[49-50];硫酸化茯苓多糖可减少H2O2的产生,保护脑神经元细胞[51];淫羊藿素可通过抗氧化而保护神经元免受MPP+诱导的神经毒性作用[52];银杏叶提取物可改善脑内OS,保护黑质DA能神经元[53]。
3.2 氧化应激通路 目前OS在PD发病机制中的作用受到越来越多的关注,了解OS 相关信号通路与PD的关系,有助于寻找新的治疗靶点。
3.2.1 Nrf2 通路 Nrf2 是参与细胞氧化还原反应调节的转录因子。Nrf2 可通过介导氧化还原途径调节抗氧化酶及基因表达,增加细胞对OS的抗性,已有研究提示其可作为PD的治疗靶点[54]。研究发现,在6-OHDA 模型中,Nrf2-抗氧化响应元件(antioxidant response element,ARE)通路对毒素诱导的PD 可发挥保护作用;OS发生后,Nrf2释放并转移到细胞核中与ARE结合,导致星形胶质细胞中的14,15-环氧化二十碳三烯酸(14,15-EET)激活Nrf2-ARE 通路发挥抗氧化作用[55]。von Otter 等[56]的研究显示,由NFE2L2基因编码的转录因子Nrf2 是细胞抵御OS 的重要调节因子,NFE2L2 蛋白变异与特发性PD 的发病机制有关。齐献忠等[57]报道,过表达含有Src同源结构域2的衔接蛋白(SHC)3能够保护PD的OS损伤,其机制与激活AKT/Nrf2/GSH 通路有关。尿酸可促进Nrf2的积累,从而抑制OS和神经元细胞死亡,对PD发挥神经保护作用[58]。
3.2.2 Toll 样受体4(Toll-like receptors 4,TLR4)通路 TLR4 在调节转录因子激活蛋白-1 (AP-1)中起重要作用;AP-1 是一种氧化还原敏感转录因子,可介导众多基因对OS等多种生理和病理刺激的反应。研究显示,在MPTP模型中TLR4-敲除(KO)小鼠DA能神经元中AP-1表达下降、ROS减少、TH阳性DA能神经元减少,可缓解PD 小鼠的运动障碍,提示TLR4在PD发病机制中起关键作用[59-61]。CYP2J蛋白存在于人类和啮齿动物大脑区域的神经细胞中,大脑中CYP2J 水平增高可延缓由炎症或神经毒素引发的PD 的病理进展;TLR4 信号通路激活可抑制大脑CYP2J 导致的星形胶质细胞中抗氧化系统的下调。以上研究提示,TLR4与OS密切相关。
4 LRRK2 基因突变与PD
LRRK2基因是PD较为常见的常染色体显性遗传致病基因。LRRK2基因突变是晚发性PD常见的遗传原因,约占PD 总数的3%。近期研究显示,部分家族性和散发性PD也与LRRK2基因突变有关(表3)。

表3 与帕金森病相关的LRRK2基因突变位点Tab.3 LRRK2 gene mutations related to Parkinson's disease
4.1LRRK2基因突变的触发 LRRK2 是一种大蛋白,由2527 个氨基酸残基组成,具有多个结构域[62]。研究发现,LRRK2 相关的发病机制涉及多个信号通路和细胞过程,如自噬、线粒体动力学等[63]。LRRK2处于关键位置,主要功能包括摄取α-syn、溶酶体加工和释放并参与致病性α-syn的传递。研究发现,LRRK2 在调节TH 纹状体通路特别是纹状体DA释放中起着至关重要的作用[64];一些受LRRK2突变影响的PD 患者不存在路易体;野生型LRRK2 可能在OS刺激下成为促凋亡因子。Mendivil-Perez等[65]报道,LRRK2突变体使OS易感性提高,导致细胞死亡增加。目前LRRK2基因突变相关PD的针对性治疗措施主要为应用LRRK2 激酶抑制剂或致力于降低LRRK2 的表达。LRRK2 激酶抑制剂(如IN-1、CZC-25146、DNL201、MLi-2 等)可通过抑制激酶的活性,终止或减缓PD患者的运动障碍和神经化学表型的进展,这些激酶抑制剂中,有多种正处于临床试验阶段[66-67]。降低LRRK2 表达的措施主要是应用反义寡核苷酸(如化合物BIIB094、ION859)绕过外周效应且阻断中枢神经系统中LRRK2 的活性,这类化合物可减少LRRK2的表达,降低靶细胞中激酶的活性,缓解DA能细胞丢失,进而改善运动障碍(图2)[68]。
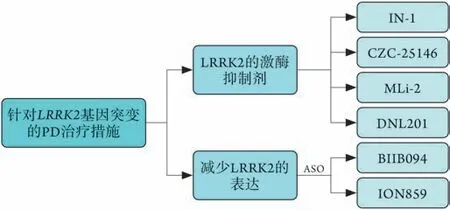
图2 针对LRRK2基因突变的帕金森病治疗措施Fig.2 The therapeutic measures to Parkinson's disease with LRRK2 gene mutation
4.2LRRK2基因突变与线粒体功能障碍 在PD 患者中已确定多种基因在线粒体稳态或线粒体自噬中发挥作用,而LRRK2可能与包含这些基因的通路子集相交。野生型LRRK2的过度表达可增强 Drp1介导的线粒体断裂。活性异常的LRRK2 也可能会聚集在线粒体,以依赖激酶的方式介导神经元毒性。研究显示,LRRK2 突变可影响线粒体的轴突运输,进而影响线粒体功能,在PD 的发病机制中起重要作用,但目前LRRK2激酶抑制剂还无法挽救这一缺陷[69-70]。在没有神经元丢失的情况下,突变的LRRK2 会损害DA神经传递,而较高水平的LRRK2表达可通过异源启动或病毒递送导致小鼠和大鼠的DA 能神经元死亡[71]。以上研究提示,由LRRK2基因突变触发的线粒体功能障碍是PD 发病的重要因素,这对探索PD的新治疗措施有重要意义。
4.3LRRK2基因突变位点 研究显示,LRRK2约有20 多个突变位点与PD 相关, 其中G2019S、N1437H、R1441C是目前研究较多的突变位点。
4.3.1 G2019S 突变 G2019S 突变相关的PD 患者发病与年龄的相关性研究显示,50岁以上的G2019S突变者PD发病率呈线性增长。2005年,p.G2019S被发现是LRRK2的常见突变,溶酶体缺陷会对细胞健康和存活产生影响,并最终导致细胞死亡,PD是这种功能障碍的结果。LRRK2G2019S 突变可增强激酶活性,进而损害自噬过程,导致α-syn在体外和体内大量积累。LRRK2G2019S 突变使溶酶体增大,导致溶酶体功能受损,且与PD 基因相关的溶酶体ATP 酶ATP13A2在LRRK2G2019S小鼠和人的脑样本中表达增多,提示与PD 相关的LRRK2 以激酶依赖的方式干扰溶酶体功能[72](表3)。多数学者认为溶酶体功能受损与多种神经退行性疾病具有一致的特征,因此如何干扰溶酶体功能并纠正这种功能障碍的发生机制,是未来相关研究的努力方向[73]。
4.3.2 N1437H 突变 研究发现,位于LRRK2 蛋白的一个复杂蛋白(ROC)GTPase 结构域内的PD 相关N1437H突变会导致其激酶域异常过度激活。目前认为ROC GTPase 结构域是理解LRRK2 功能及其致病机制的关键。大多数PD 相关的LRRK2 突变可导致激酶活性增高,提示这种过度激活与PD的发病机制有关[74]。2004年,安德烈亚斯·普施曼等对N1437H突变的PD患者进行了首次脑DNA神经病理学研究,结果显示黑质中的细胞几乎完全丢失,且脑干中有α-syn阳性病变,皮质中存在少量的路易体[75-76](表3)。
4.3.3R1441C突变 在与PD相关的LRRK2突变中,R1441C 突变的发生率仅次于G2019S 突变。2004 年的研究发现,携带R1441C 突变的PD 患者病理学表现为黑质中DA 能细胞死亡。研究发现,R1441C 转基因小鼠出现晚发性DA能神经元死亡和运动功能障碍等PD表型[77];p.R1441C携带者、p.G2019S携带者和散发性PD之间的临床表现具有相似性,如发病年龄分布、临床特征等[78](表3)。
5 总结与展望
综上所述,导致DA 能神经元变性的α-syn、线粒体功能障碍、OS、LRRK2基因突变等因素均与PD的发病有密切联系。目前对于PD的发生机制研究十分广泛,取得了很大进展,但相关的临床研究还十分匮乏。当前PD的治疗以对症治疗为主,面对不同的治疗靶点往往需要具体问题具体分析。例如,以α-syn为靶点治疗PD时,主要措施是抑制α-syn的聚集、传递及其异常折叠,进而促进α-syn消除;以线粒体功能障碍为治疗目标时,主要措施是恢复线粒体功能、增加线粒体的生物活性;专注于抗氧化治疗时,主要措施是抑制氧化损伤和抑制细胞凋亡;以LRRK2 为靶点治疗PD 时,主要措施是抑制LRRK2 激酶活性或减少LRRK2 的表达。然而研究显示,PD的多种致病因素往往相互作用、相互联系而共同致病,使相关研究的难度增大。因此,针对PD作用机制和治疗的研究不能仅着眼于单个靶点。PD治疗药物的研发需要更多地关注于多途径、多靶点的研究,如筛选既可抑制氧化应激、也能保护线粒体功能的化合物等。因此,探明PD的发病机制,研究如何阻止黑质DA 能神经元变性,对于PD 治疗研究的进一步推进有重要意义。
