脓毒症相关急性肾损伤的病理生理机制
2023-09-19栗小茹综述李世军审校
栗小茹 综述 李世军 审校
脓毒症指机体对感染反应失调引起的危及生命的器官功能障碍综合征,急性肾损伤(AKI)是其最常见的并发症。脓毒症相关急性肾损伤(SA-AKI)占危重症AKI病例的45%~70%[1],病死率高,预后差,严重危害患者生命。其包含由脓毒症直接导致的AKI和脓毒症相关因素(如肾毒性药物、容量状态、腹腔间隔室综合征等)间接导致的AKI。近期,《第28届急性疾病质量倡议(ADQI)工作组共识报告》提议将脓毒症诊断后7 d内发生的AKI定义为SA-AKI,7 d后发生的AKI可能与初始脓毒症损伤无直接关系[2]。
SA-AKI的发病机制复杂,尚未完全阐明,有多种因素参与(图1),表现为多种病理生理机制同时存在,背景易感因素(如合并症、基因和表观遗传等)及耐受力(即宿主减弱组织损伤的能力)之间的复杂相互作用。临床表型也存在差异,来自早期感染性休克流程化治疗(ProCESS)队列的数据表明,处于改善全球肾脏病预后(KDIGO)标准定义的AKI同一分期的患者,损伤生物标志物阳性的AKI相比损伤生物标志物阴性的AKI,30 d存活率更低[3]。目前尚未有确切、有效的预防和治疗手段。本文拟就SA-AKI的病理生理机制研究进展作一综述。
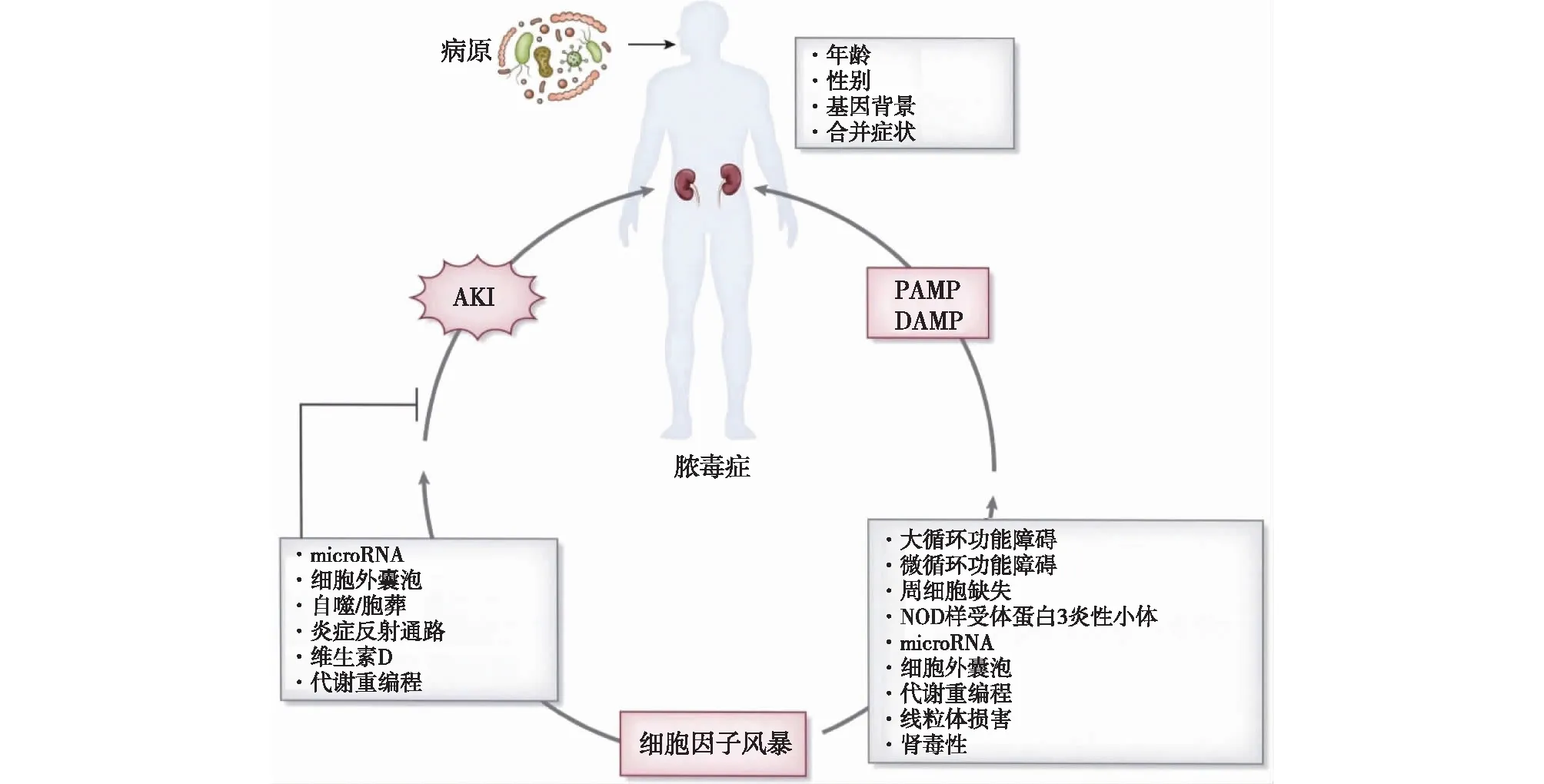
图1 SA-AKI的发病机制[4]
微循环障碍
既往认为缺血再灌注损伤导致急性肾小管坏死是SA-AKI发生的主要机制,逐渐有证据表明,在没有肾脏低灌注,血流动力学稳定,甚至全肾血流量正常或增加的情况下,也可以发生SA-AKI,说明微循环功能障碍可能在肾损伤的发展中起重要作用。多数研究发现,脓毒症期间管周毛细血管微循环发生明显的异质性改变,表现为有持续血流血管的比例减少,间断血流和无血流血管的比例增加。Sun等[5]通过实时测量脓毒症小鼠的微血流动力学和氧代谢变化发现,脓毒症导致肾小管周围毛细血管氧饱和度急剧下降,且伴随肾脏ATP水平的显著降低,而管周毛细血管血流和血清肌酐仅轻微改变。表明脓毒症期间肾脏微循环功能障碍、局部血流淤滞伴随氧代谢的变化促进了AKI的发生。此外,肾脏微血管内皮细胞损伤、管周细胞损伤缺失参与微循环功能障碍的形成。
内皮细胞损伤血管内皮细胞最早感知毒素刺激,其作为炎症反应和组织损伤的启动部位,首先与循环中损伤相关分子模式(DAMP)、病原体相关分子模式(PAMP)结合,激活细胞内信号转导使肾脏血管内皮细胞表型发生改变,转变为促炎和分泌表型,可启动细胞内信号和炎症介质转录,释放组织因子和促炎细胞因子等进入循环,放大肾脏炎症损伤;转变为促黏附表型,可释放细胞间黏附分子和选择素分子,与循环的白细胞结合,促进白细胞向肾间质浸润;转变为促凝表型,可导致血小板的募集和凝血的激活,从而导致肾脏微血管血栓形成和损伤。Star等[6]通过对早期脓毒症患者血浆成分进行靶向蛋白质组学分析,发现53种蛋白质与AKI发生相关,进一步行富集通路分析发现,富集的前3条通路与血管内皮调节相关,其分别是血管内皮生长因子受体(VEGFR)1和VEGFR2介导的信号通路、血管生成素受体Tie2介导的信号通路、血小板源性生长因子(PDGF)受体信号通路,说明肾脏血管内皮细胞功能障碍可能先于AKI的发生,提示脓毒症早期血管内皮细胞保护策略可能有助于防止SA-AKI的进展。
周细胞损伤缺失周细胞间断包绕在肾小管管周毛细血管管腔外侧,作为支持细胞,具有稳定血管、控制张力、维持管周毛细血管微环境的作用。研究证实周细胞是慢性肾脏病(CKD)肾脏纤维化中肌成纤维细胞的主要来源。周细胞通过特定的黏附点、黏附斑、缝隙连接和紧密连接与毛细血管内皮细胞相互作用,进而增强内皮细胞屏障功能。Zhang等[7]在脓毒症小鼠中发现,脓毒症时,周细胞脱落、内皮细胞鞘氨醇-1-磷酸受体(S1PR)表达增加,钙黏蛋白等水平下降,继而血管内皮屏障破坏及血管低反应性发生。Castellano等[8]在脂多糖(LPS)诱导的猪AKI模型中发现,注入LPS 9 h后,即可观察到周细胞向肌成纤维细胞转分化,进而促进肾功能的丢失。因此,深入探索周细胞与脓毒症微循环障碍的关系将有助于进一步认识SA-AKI的机制。
线粒体功能障碍
肾小管上皮细胞(RTEC)富含线粒体,以维持肾小管对物质重吸收和分秘过程的巨大耗能。SA-AKI早期即有线粒体能量代谢紊乱及功能障碍的发生。代谢重编程指细胞代谢从氧化磷酸化(OXPHOS)转变为糖酵解供能。脓毒症早期短时间内,细胞能量代谢切换为有氧糖酵解可在一定程度上缓解能量危机,优化能量供应以维持重要生命活动,确保细胞短期存活,是一种自我耐受机制。一方面,有氧糖酵解使OXPHOS和线粒体活性氧(ROS)产生减少,保护细胞和线粒体免受进一步氧化损伤。另一方面,与氧化磷酸化相比,糖酵解可更快速率产生ATP和代谢合成原料,有助于免疫细胞快速活化、增殖而发挥功能。但是大量研究表明,过度糖酵解也会损害器官细胞功能,Tan等[9]在脓毒症小鼠和LPS处理的HK-2细胞中观察到,糖酵解代谢产物乳酸可下调SIRT3和磷酸化AMP活化蛋白激酶(p-AMPK)的表达,抑制自噬并促进细胞凋亡,而有氧糖酵解抑制剂2-脱氧-D-葡萄糖(2-DG)处理后看到相反的结果,改善了脓毒症引起的肾损伤。另外过度糖酵解也会导致免疫抑制,高水平乳酸可通过多种机制抑制M1型巨噬细胞促炎活性,并促进M2型巨噬细胞发挥过度抗炎作用[10]。因此,糖酵解及时转换为OXPHOS对于恢复细胞功能非常必要,而SA-AKI期间代谢重编程的灵活转化以及代谢重编程和各种细胞功能之间的相互作用仍然需要深入探索。
脓毒症可损害线粒体质量控制机制,如生物发生,融合与裂解,线粒体自噬等,导致线粒体功能障碍。PGC-1α是线粒体生物发生重要的调节因子,脓毒症时肾小管上皮细胞PGC-1α的表达降低,生物合成减少,抑制线粒体功能的恢复,进而影响肾小管功能。通过诱导pGC-1α可促进AKI改善,如烟酰胺腺嘌呤二核苷酸(NAD+)可激活Sirt 1的活性,靶向pGC-1α,当补充NAD+前体烟酰胺单核苷酸或NAM,有助于顺铂或缺血诱导的AKI的肾脏保护[11]。线粒体融合与裂解依赖多个信号分子调节,如线粒体融合蛋白(Mfn)、视神经萎缩蛋白(OPA1)和Dynamin相关蛋白1(Drp1)等。脓毒症时,线粒体分裂过度或融合受阻可使线粒体碎片化,导致线粒体肿胀破裂、ROS激活及线粒体膜通透性改变,促进肾小管损伤。SA-AKI早期,线粒体自噬可清除RTEC中受损的线粒体,减少氧化应激和凋亡,发挥肾脏保护作用。但在后期,完整的自噬体无法形成从而导致受损的线粒体在细胞中积聚,加重肾脏损害。目前已知一些针对线粒体的治疗靶点(如腺苷酸活化蛋白激酶、Drp1、Mfn-2、ROS、自噬相关基因Ambral等)已取得一定进展[12],进一步探索线粒体相关靶点,有望实现SA-AKI的早期干预。
免疫功能紊乱
机体感染病原体后,DAMP或PAMP可与免疫细胞、血管内皮细胞、肾小管上皮细胞表面或胞质内的模式识别受体[如Toll样受体(TLR)、NOD样受体(NLR)和视黄酸诱导基因蛋白-I(RIG-I)样受体]结合,启动下游信号通路,导致大量炎症介质(如血管活性物质、细胞因子、趋化因子、氧自由基、急性时相反应物质等)释放,进一步激活细胞固有免疫和适应性免疫系统、补体系统、凝血系统,诱发细胞因子风暴,使组织损伤放大,致使AKI发生。大量研究表明,免疫细胞与肾组织损伤、预后及转归紧密相关。自然杀伤(NK)细胞具有细胞毒功能,可释放细胞毒介质(如穿孔素FasL和颗粒酶)或产生促炎因子(干扰素γ)介导肾血管内皮和小管上皮细胞损伤,Uchida等[13]在接受持续肾脏替代治疗的SA-AKI患者中发现,CD56+NK细胞和CD56+T细胞(NKT)上的穿孔素表达增加及NKT细胞上的FasL表达上调,说明穿孔素和FasL通路可能与SA-AKI的发病相关。尿激酶型纤溶酶原激活剂受体(uPAR)是一种糖基化磷脂酰肌醇锚定蛋白,在细菌感染后表达上调,其主要表达于单核细胞和中性粒细胞等固有免疫细胞,与炎症反应、免疫细胞激活和迁移有关。uPAR通过裂解膜结合形式产生可溶性uPAR(suPAR)。Nusshag等[14]分析了脓毒症患者血清suPAR水平与AKI严重程度的关系,发现suPAR水平>12.7 ng/mL的患者发生肾脏替代治疗或死亡的风险最高,调整后的比值比为7.48(95%CI 3.00~18.63)。同样,他们通过建立多菌脓毒症小鼠模型,发现suPAR高表达的小鼠肾脏损伤更重、存活率更低,肾脏免疫荧光染色显示皮质和髓质中有显著的CD4+和CD8+细胞聚集。而uPAR基因敲除小鼠表现出强大的保护作用,包括肾功能和存活改善。以上说明,免疫反应过程中潜在的病理生理学驱动因素可能对寻找SA-AKI的诊断、预后生物标志物及治疗靶点有重要意义。
随着时间推移,SA-AKI患者可迅速出现严重的免疫抑制,表现为免疫效应细胞增殖和功能下降,单核细胞功能障碍,T辅助细胞(Th)免疫表型向Th2表型转变,调节性T细胞(Treg)扩增,抗炎介质释放增加,细胞凋亡增加,同时,人类白细胞抗原-DR(HLA-DR)的表达减少及免疫检查点分子——如T细胞表面的程序性死亡受体1(PD-1)、T淋巴细胞免疫球蛋白黏蛋白3(TIM-3)等——表达增加会进一步加重免疫抑制。这些导致宿主无法根除原发感染,继发感染概率增加,与患者的预后不良和死亡增加相关。Xu等[15]在盲肠结扎穿孔诱导的脓毒症小鼠AKI模型中发现,SA-AKI存在肾小管上皮细胞程序性死亡配体1(PD-L1)过度表达和CD3+T淋巴细胞减少,此外脓毒症时高乳酸水平可上调PD-1/PD-L1通路,诱导SA-AKI中淋巴细胞凋亡而导致免疫抑制。
NOD样受体蛋白3炎性小体(NLRP3) NLRP3是位于细胞质中一种模式识别受体(PRR),在SA-AKI早期固有免疫和炎症级联释放中充当重要的中间介质。PAMP、DAMP或细胞因子[如白细胞介素(IL)-1β和肿瘤坏死因子α(TNF-α)]作为启动信号,可以促进细胞核中NLRP3、促IL-1β和促IL-18基因表达。另一方面,线粒体ROS产生、细胞内低钾浓度或组织蛋白酶B释放等多种信号可触发NLRP3激活,使下游caspase-1生成和活化,促进IL-1β和IL-18的成熟与释放,进而导致肾脏炎症反应和肾小管损伤。目前研究显示,在AKI的发生发展过程中,NLPR3可以是多条通路的下游靶点(如P2X7R/TLR/MAPK/mROS-TXNIP/NF-κB/DAPK-NLRP3),激活后发生正性调控进而发挥促炎和促纤维化效应[16]。多种化学成分或药物可作用于相关通路,抑制NLPR3的激活从而减轻脓毒症AKI,如苦参碱通过调节PTPN2/JNK/SREBP2通路抑制脓毒症NLRP3激活,可有效缓解LPS诱导的小鼠脓毒症症状[17]。右美托咪啶可通过α2-AR/AMPK/mTOR途径增强自噬后抑制NLRP3炎性小体的激活,进而缓解LPS诱导小鼠的肾脏损伤[18]。这些说明抑制NLPR3的激活很有可能成为SA-AKI的有效干预靶点。
microRNA(miRNA) miRNA属于非编码RNA家族,作为一种基因调控分子主要参与调节转录后翻译及基因表达的表观修饰。已知SA-AKI中存在多种miRNA上调或者具有保护作用的miRNA下调,通过靶向相关基因或信号通路(如NF-κB、PTEN和JNK等),促进细胞炎症、凋亡、焦亡、自噬和氧化应激,加重肾脏损伤[19]。Ji等[20]证明在SA-AKI患儿血清中miR-320-3p水平显著升高,结合中性粒细胞明胶酶相关脂质运载蛋白(NGAL)、肾损伤分子1(KIM-1)和急性生理和慢性健康评估(APACHE) Ⅱ评分可有效预测患儿预后。因而miRNA在SA-AKI的靶向治疗、早期预测及评估预后方面有极大的应用价值。
细胞外囊泡(EV)EV是由细胞释放的脂质双分子层包裹的微粒,里面包含DNA、RNA、miRNA、蛋白、脂质等多种分子和其他细胞质成分,其携带的分子在细胞间转移以传递信号、调节靶细胞基因组,并介导多种旁分泌和内分泌作用调节局部或远处宿主细胞。EV通过发挥来源细胞(如内皮细胞、血小板、小管上皮细胞、巨噬细胞和其他免疫细胞等)的表型作用,或本身作为毒性循环介质,在血管内皮损伤、微血管血栓、炎症凋亡、巨噬细胞极化、氧化应激、线粒体损伤等方面介导肾脏损伤,促进SA-AKI的发生。Xiang等[21]近期在体内外实验中研究发现,在SA-AKI中,肾小球巨噬细胞浸润及外泌体分泌增加,介导肾小球内皮细胞损伤,促进AKI进展。酸性鞘磷脂酶(ASM)是一种表达于溶酶体表面的水解酶,可将神经鞘蛋白水解成神经酰胺,进而在细胞膜表面形成一个弯曲的浓缩平台,以促进外泌体运输。研究发现ASM可调节巨噬细胞外泌体的分泌,而抑制巨噬细胞ASM可减少内毒素刺激后外泌体的释放,表现出对肾脏的保护作用,表明靶向EV及ASM可能是SA-AKI潜在的治疗靶点。此外,Liu等[22]在SA-AKI小鼠模型和HK-2细胞中发现,由miR-342-5p修饰脂肪来源的间充质干细胞分泌的外泌体通过转移miR-342-5p,可以抑制TLR9进而加速自噬,减轻AKI,说明干细胞来源的EV以及工程修饰的EV有望成为治疗SA-AKI的有力手段。
自噬在应激诱导(如ROS、内质网应激、缺氧,DNA损伤与免疫信号传导等)下,细胞可通过自噬降解受损的细胞器和大分子,以维持细胞内稳态。自噬激活存在于各种形式AKI中,是肾小管上皮细胞抵抗损伤和死亡的防御机制。SA-AKI中自噬发生动态变化,Sun等[23]在CLP和LPS诱导的AKI小鼠模型及HK-2细胞中观察到,毒素刺激后RTEC的自噬水平一过性升高,随后急剧下降,且自噬抑制伴随着肾小管损伤组织学评分(基于肾小管刷状缘消失、肾小管出血、肾小管管型、炎性细胞浸润、皮质明显坏死五个方面评估)的增加,而自噬激动剂可减少脓毒症后肾小管的损害,说明自噬激活对SA-AKI发挥保护作用。研究还发现,白藜芦醇/槲皮素通过激活去乙酰化酶Sirt1或p53乙酰化赖氨酸位点突变,诱导p53去乙酰化,可以促进RTEC自噬,减轻SA-AKI。同样有证据表明,右美托咪定可以通过PI3K/AKT/mTOR途径激活自噬,以防止脓毒症诱导的肾损伤[24]。因此,通过药物诱导自噬可能是SA-AKI潜在的治疗方式。
小结:SA-AKI发病机制复杂,是多种病理生理机制、背景易感因素和组织耐受能力之间相互作用的结果。SA-AKI在临床表型、疾病进展、治疗反应等多方面存在异质性,基于尿量和血清肌酐定义的临床表型难以追溯到特定的病理生理机制。如何将临床前模型中识别的机制应用于临床,利用生物标志物将临床表型与特定病理生理机制相联系,优化治疗提高治疗有效性有待进一步探索。
