和厚朴酚自乳化释药系统的制备与药动学评价
2023-09-13王萌
王 萌
咸阳职业技术学院,咸阳 712000
和厚朴酚(honokiol,HK)是从木兰科落叶乔木植物厚朴的干皮、根皮及枝皮中分离得到的一种天然化合物,具有抗炎[1]、抗氧化、抗衰老[2]等药理活性。近年来HK的抗肿瘤活性受到了广泛关注,其对肺癌、前列腺癌、乳腺癌和肝癌等具有抑制作用[3],但极低的溶解度[4]和口服生物利用度[5]严重影响了其药用价值,因此,有必要设计合适的药物递送系统来提高HK的溶解度,进而提高其生物利用度和治疗效果[6]。自乳化释药系统(self-emulsifying drug deli-very systems,SEDDSs)是由药物、油、表面活性剂和助表面活性剂构成的各相同性混合物,其在水性介质中温和搅拌下自发形成纳米级水包油(O/W)型乳液[7],能够避免药物的首过效应[8],抑制由P-糖蛋白介导的外排作用[9],并且能够增强药物的渗透性[10],在提高难溶性药物的口服生物利用度方面具有广阔的应用前景[11]。本研究制备和厚朴酚自乳化释药系统(HK-SEDDSs),并通过大鼠体内药动学评估药物的生物利用度,为提高HK的口服生物利用度提供一种有效的方案。
1 仪器与材料
1.1 仪器
HSY-B型智能水浴恒温振荡器(常州申光仪器有限公司);Zetasizer Nano ZS型激光粒度仪(英国马尔文公司);H1750型台式高速离心机(湖南湘仪实验室仪器开发有限公司);JEM-F200型透射电镜(日本电子株式会社);RC-8DA型药物溶出仪(天津创兴电子设备制造股份有限公司)。
1.2 试药
和厚朴酚(广州合诚三先生物科技有限公司);橄榄油(olive oil)、玉米油(corn oil)、聚氧乙烯蓖麻油(cremophor EL 35)、聚氧乙烯氢化蓖麻油(cremophor RH 40)均由德国巴斯夫辅料公司提供;辛酸/癸酸三酰甘油(miglyol 812)、单辛酸甘油酯(capmul MCM C8)、二乙二醇单乙基醚(transcutol P)、辛酸癸酸聚乙二醇甘油酯(labrasol)、丙二醇单辛酸酯(capryol 90)均由法国嘉法狮公司提供;吐温80(tween 80)、聚乙二醇200(PEG200)均由南京威尔药业集团股份有限公司提供。
1.3 动物
Wistar大鼠,雌雄各半,2~3月龄,体质量为(200~250) g,购自第四军医大学实验动物中心,生产许可证号SCXK-(军)2012-0007,质量合格证号20150021673。
2 方法与结果
2.1 平衡溶解度的测定
用摇瓶法测定HK在不同油相、表面活性剂和助表面活性剂中的溶解度[12]。于若干个干净的玻璃小瓶中,分别加入待筛选的油、表面活性剂和助表面活性剂各1.5 mL,再加入过量的HK原料药,加盖密封,固定于水浴摇床中,(37±0.5) ℃持续振摇72 h,以10 000 r·min-1离心5 min,收集上清液,适当稀释后用高效液相色谱(HPLC)法检测每个组分中HK的含量,计算平衡溶解度。结果见表1。
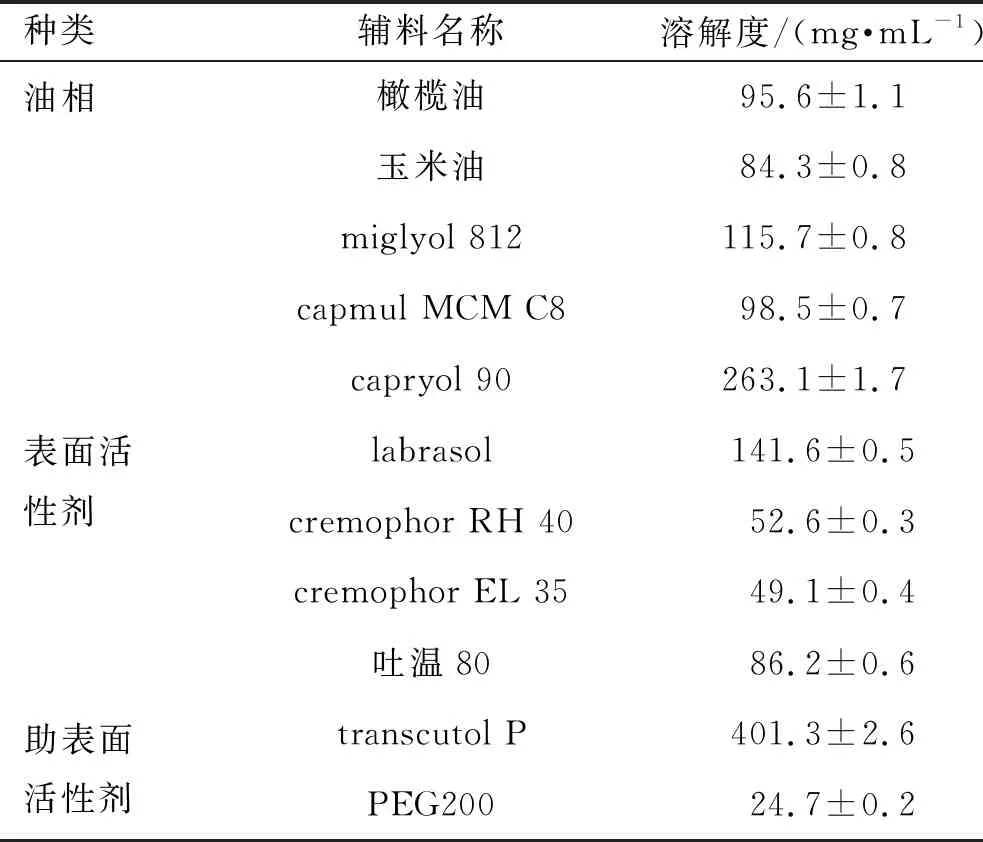
表1 和厚朴酚在不同辅料中的溶解度
由表1可见,HK在不同油中的溶解度顺序为capryol 90>miglyol 812>capmul MCM C8>橄榄油>玉米油,在不同表面活性剂中溶解度的顺序为labrasol>吐温80>cremophor RH 40>cremophor EL 35,在不同助表面活性剂中溶解度的顺序为trans-cutol P>PEG200。因此,本研究分别选择capryol 90、labrasol和transcutol P作为HK-SEDDSs的油相、表面活性剂和助表面活性剂。
2.2 伪三元相图的绘制
用水滴定法构建伪三元相图以确定SEDDSs的最佳自乳化区域[13]。按照表面活性剂与助表面活性剂质量比为1∶1、1∶2、2∶1配制成表面活性剂混合物(Smix),再将Smix与油相以9∶1至1∶9的比例混合均匀,取各混合液1 mL,在磁力搅拌(50 r·min-1)下缓慢滴加0.1 mol·L-1盐酸溶液(37±0.5) °C,观察出现浑浊时各处方中油、Smix和水的用量,在伪三元相图上标出能够形成微乳的区域。结果见图1。自乳化区域面积越大表示自乳化能力越强[14]。

注:A.1∶1;B.1∶2;C.2∶1。O.油相;S.表面活性剂;Co-S.助表面活性剂。
由图1可见,表面活性剂和助表面活性剂的比例为2∶1(图1C)时形成的自乳化区域的面积最大。因此,以油相capryol 90的比例为5%~25%,表面活性剂labrasol的比例为35%~75%,助表面活性剂transcutol P的比例为10%~50%,进一步优化HK-SEDDSs的处方。
2.3 HK-SEDDSs的制备
HK-SEDDSs处方中capryol 90、labrasol和transcutol P用量考察范围见表2。根据表3中的处方组成,将capryol 90、labrasol和transcutol P加入玻璃小瓶中,涡旋混合形成均匀透明的SEDDSs,再将HK原料药加入上述SEDDSs中,涡旋混合使药物完全溶解,最终得到不同处方的HK-SEDDSs。

表2 HK-SEDDSs的自变量与因变量

表3 HK-SEDDSs的处方组成及对应测定结果
2.4 质量评价
2.4.1透射率的测定 取不同处方的HK-SEDDSs各0.5 g,加入0.1 mol·L-1盐酸溶液稀释100倍,形成微乳,用紫外可见分光光度计测定各样品在638 nm处的透光率,通过透光率可初步判断形成微乳粒径的大小。
2.4.2自乳化时间的测定 于溶出杯中加入0.1 mol·L-1盐酸溶液500 mL,温度为(37±0.5) ℃,转速为50 r·min-1,取不同处方的HK-SEDDSs各0.5 g,置于溶出杯中,肉眼观察每组样品自乳化形成微乳的时间,并做记录。自乳化时间越短,表示自乳化能力越强。
2.4.3粒径分布的测定 取不同处方的HK-SEDDSs各0.5 g,加入0.1 mol·L-1盐酸溶液稀释100倍,形成微乳,取上述样品加入聚苯乙烯样品池中,用Zetasizer Nano ZS型激光粒度仪测定粒度分布情况。
2.5 Box-Behnken实验设计
根据伪三元相图结果,以油相capryol 90的比例为5%~25%(x1)、表面活性剂labrasol的比例为35%~75%(x2)、助表面活性剂transcutol P的比例为10%~50%(x3)作为自变量,制备HK-SEDDSs,以HK-SEDDSs的透光率(y1)、自乳化时间(y2)和粒径分布(y3)作为评价指标。因素与水平见表2。用3因素3水平Box-Behnken实验设计对其处方进行研究[15],共进行17次实验,结果见表3。所得的实验数据使用方差分析、多元二次方程和效应面图进行评价。capryol 90、labrasol和transcutol P不同比例对HK-SEDDSs透光率影响的方差分析结果见表4。capryol 90,labrasol和transcutol P不同比例对HK-SEDDSs的透光率的效应面图见图2。capryol 90、labrasol和transcutol P不同比例对HK-SEDDSs自乳化时间影响的方差分析结果见表5。capryol 90、labrasol和transcutol P不同比例对HK-SEDDSs的自乳化时间影响的效应面图见图3。capryol 90、labrasol和transcutol P不同比例对HK-SEDDSs粒径分布影响的方差分析结果见表6。capryol 90、labrasol和transcutol P不同比例对HK-SEDDSs粒径分布影响的效应面图见图4。
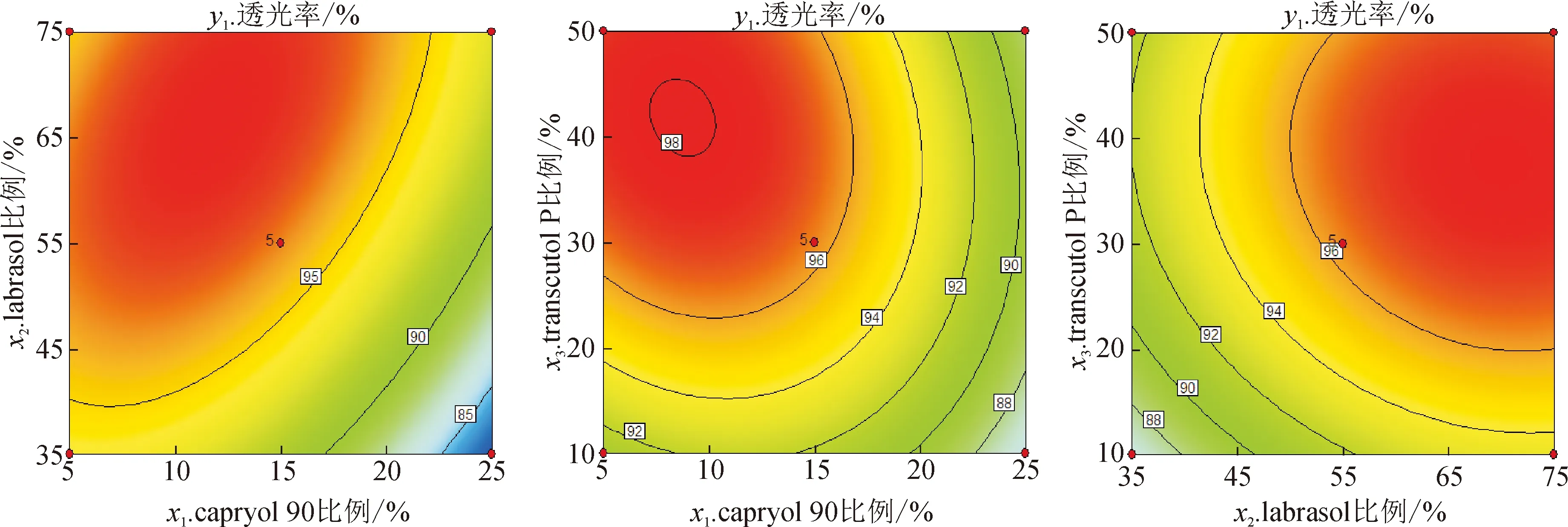
图2 Capryol 90 (x1)、labrasol (x2)和transcutol P (x3)不同比例对HK-SEDDSs透光率 (y1)影响的效应面图

图4 Capryol 90 (x1)、labrasol (x2)和transcutol P (x3)不同比例对HK-SEDDSs粒径分布(y3)影响的效应面图
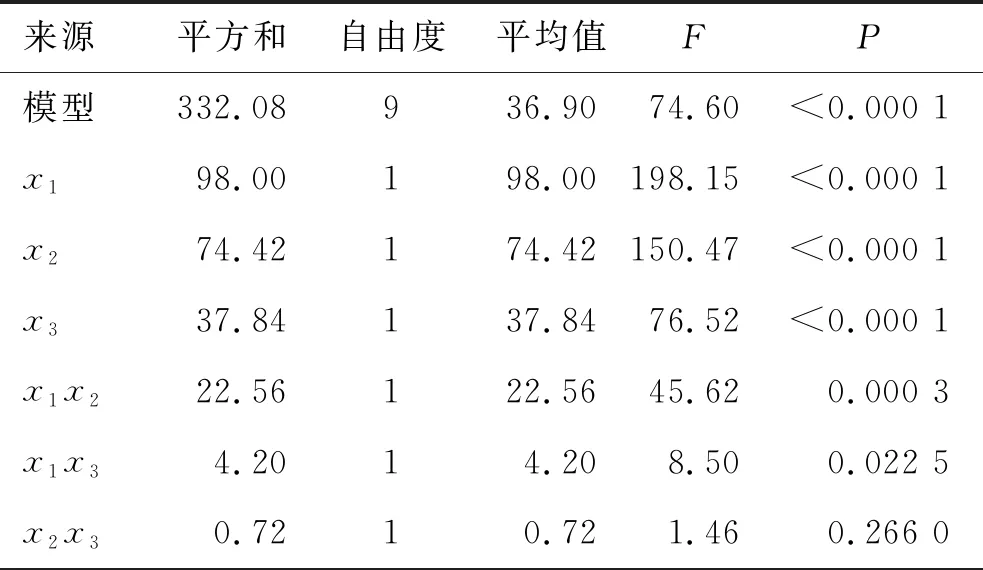
表4 capryol 90(x1)、labrasol(x2)和transcutol P(x3)不同比例对HK-SEDDSs透光率(y1)影响的方差分析结果

表5 Capryol 90(x1)、labrasol(x2)和transcutol P(x3)不同比例对HK-SEDDSs自乳化时间(y2)影响的方差分析结果
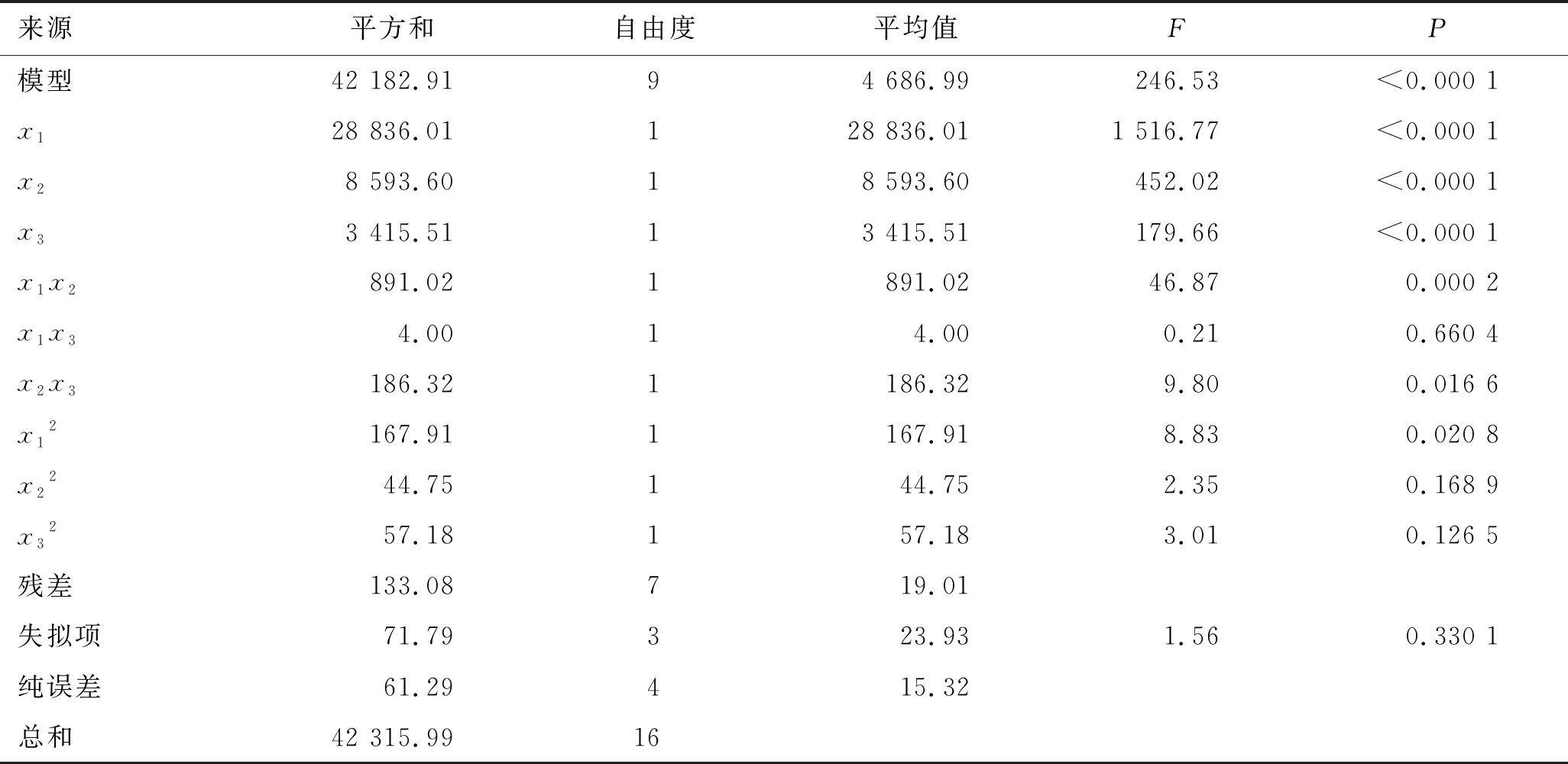
表6 capryol 90 (x1)、labrasol(x2)和transcutol P(x3)不同比例对HK-SEDDSs粒径分布(y3)影响的方差分析结果
所制备的17组HK-SEDDSs样品的透射率在82.4%~98.1%范围内,由表4可见,capryol 90的比例(x1)、labrasol的比例(x2)和transcutol P的比例(x3)对HK-SEDDSs的透光率(y1)均具有显著影响(P<0.05)。由图2可见,随着处方中油相capryol 90比例的上升,HK-SEDDSs的透光率增大,这是由于表面活性剂和助表面活性剂的量不足造成的;而随着处方中表面活性剂labrasol和助表面活性剂transcutol P比例的上升,HK-SEDDSs的透光率增大,这是由于表面活性剂和辅助表面活性剂共同作用降低了油水界面的张力,使形成的微乳的粒径更小,提高了透光率。使用多元二次方程对3个变量与透光率(y1)之间的关系进行方程拟合:y1=96.34-3.50x1+3.05x2+2.17x3+2.38x1x2-1.02x1x3-0.42x2x3-3.28x12-1.93x22-2.38x32(R2=0.989 7)。
所制备的17组HK-SEDDSs样品的自乳化时间在23~147 s范围内。由表5可见,capryol 90比例(x1)、labrasol比例(x2)和transcutol P比例(x3)对HK-SEDDSs的自乳化时间(y2)均具有显著影响(P<0.05)。由图3可见,随着处方中油相capryol 90比例的上升,HK-SEDDSs的自乳化时间有所延长,而随着处方中表面活性剂labrasol比例和助表面活性剂transcutol P比例的上升,HK-SEDDSs的自乳化时间缩短,这是由于表面活性剂和辅助表面活性剂共同作用降低了油水界面的张力,提高了自乳化性能。3个变量与自乳化时间(y2)之间的二次多项式拟合方程为y2=63.20+30.62x1-19.75x2-23.13x3-13.25x1x2-9.00x1x3+14.25x2x3+22.40x12-2.35x22-7.60x32(R2=0.999 6)。
所制备的17组HK-SEDDSs样品形成的微乳粒径分布在29.5~214.6 nm范围内。由表6可见,capryol 90比例(x1)、labrasol比例(x2)和transcutol P比例(x3)对HK-SEDDSs形成的微乳粒径的分布(y3)均具有显著影响(P<0.05)。由图4可见,随着处方中油相capryol 90比例的上升,微乳的粒径增大;随着处方中表面活性剂labrasol比例和助表面活性剂transcutol P比例的上升,微乳的粒径减小。3个变量与粒径分布(y1)之间的二次多项式拟合方程为y3=113.08+60.04x1-32.77x2-20.66x3-14.93x1x2+1.00x1x3+6.83x2x3-6.32x12+3.26x22+3.69x32(R2=0.996 9)。
HK-SEDDSs口服进入体内,要求在短时间内迅速自乳化形成粒径较小的微乳,这样有利于药物的充分吸收与利用[16],因此本研究要求制备的HK-SEDDSs的透光率最大化,自乳化时间最短化,形成的微乳粒径分布最小化。经实验软件优化得到HK-SEDDSs的最佳处方为capryol 90、labrasol和trans-cutol P的质量比为10∶60∶30。
2.6 HK-SEDDSs性质研究
2.6.1热力学稳定性 本研究用离心、低温-高温循环、冷冻-高温循环评价HK-SEDDSs的热力学稳定性[17]。①离心考察:取HK-SEDDSs 0.5 g,加入0.1 mol·L-1盐酸溶液稀释100倍,以10 000 r·min-1离心20 min,肉眼观察是否存在相分离和相转变现象。②低温-高温循环考察:取HK-SEDDSs 1 g置于西林瓶中,在5 ℃放置48 h,取出后放置在40 ℃下48 h,连续进行3次循环,肉眼观察是否存在相分离和相转变现象。③冷冻-高温循环考察:取HK-SEDDSs 1 g置于西林瓶中,在-20 ℃放置48 h,取出后在40 ℃放置48 h,连续进行3次循环,肉眼观察是否存在相分离和相转变现象。实验结果显示,HK-SEDDSs经离心、低温-高温循环和冷冻-高温循环,均未出现相分离和相转变现象,表明HK-SEDDSs的热力学稳定性良好。
2.6.2微观形态观察 取HK-SEDDSs 0.5 g,加入0.1 mol·L-1盐酸溶液稀释100倍,形成微乳,将1滴微乳液置于碳涂层铜网上,均匀铺展,再将1滴磷钨酸滴加到样品表面,等待10 min,用滤纸在边缘吸收多余水分,置于阴凉处风干。将样品放置在透射电子显微镜下观察微乳的微观形态,见图5。由图5可见,HK-SEDDSs形成的微乳以圆球形均匀分散,其粒径大部分在30~80 nm范围内。

图5 HK-SEDDSs形成的微乳的透射电镜照片
2.7 药物溶出研究
用桨法比较HK-SEDDSs与HK原料药的体外溶出速率,以0.1 mol·L-1盐酸溶液作为溶出介质,取介质500 mL置于溶出杯中,温度为(37±0.5) ℃;将HK-SEDDSs置于透析袋(截留相对分子质量为12 000)中,系住两端,固定在搅拌桨上,开启搅拌桨,转速为100 r·min-1,在预设时间间隔(10、20、30、45、60、90、120 min)吸取5 mL溶出介质,经0.22 μm微孔滤膜过滤,进样检测药物含量,计算药物累积溶出度。另取HK原料药直接置于溶出杯中,操作同上。比较HK-SEDDSs与HK原料药的体外溶出速率,结果见图6。由图6可见,HK-SEDDSs中药物的释放速度较快,在10 min内超过60%的药物溶出,在30 min内药物基本完全溶出。相比之下,HK原料药的溶出速率较为缓慢,在120 min内仅有约25%的药物溶出。

图6 HK-SEDDSs与HK原料药在pH 1.2盐酸溶液中的溶出曲线
2.8 生物利用度研究
用Wistar大鼠(200~250 g)研究HK-SEDDSs的体内药动学行为。实验前大鼠禁食12 h,可自由饮水。将大鼠分为2组,第1组通过灌胃针口服给予HK混悬液,第2组通过灌胃针口服给予HK-SEDDSs,给药剂量均为50 mg·kg-1。口服给药后,在不同时间点(15、30 min,1、2、4、6、8、12 h)经眼眶静脉丛取血样,置于预肝素化离心管中,以4 000 r·min-1离心15 min,分离上层血浆,置于-20 °C冰箱中保存。取血浆样品200 μL,加入紫杉醇溶液50 μL作为内标,涡旋混合5 min,加入乙腈500 μL沉淀蛋白,以12 000 r·min-1离心10 min,分离有机相至尖底离心管中,减压干燥,残留物用200 μL乙腈溶解,用HPLC法分析血浆样品中药物的质量浓度[18]。色谱条件:色谱柱为C18柱(250 mm×4.5 mm,5 μm);流动相为10 mmol·L-1磷酸盐缓冲液(pH 4.5)-乙腈(80∶20);检测波长为225 nm;进样量为20 μL。使用DAS 2.0药动学软件计算达峰时间(tmax)、达峰浓度(cmax)、半衰期(t1/2)、血药质量浓度-时间曲线下面积(AUC0→∞)等药动学参数。结果见表7。
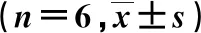
表7 HK混悬剂和HK-SEDDSs经口服给药后在大鼠体内的药动学参数
由表7可见,HK混悬剂和HK-SEDDSs的tmax分别为(1.1±0.6)、(0.7±0.4) h,cmax分别为(395.3±35.7)、(633.6±54.2) ng·mL-1,t1/2分别为(8.3±1.6)、(8.0±1.3) h,AUC0→∞分别为(1 478.8±105.3)、(2 531.6±174.3) ng·mL-1·h-1;与HK混悬剂组比较,HK-SEDDSs中HK的相对生物利用度达到了159.0%。
3 讨论
本研究通过测定HK在各种辅料中的溶解度,确定了处方中油相、表面活性剂和助表面活性剂的种类,进一步采用水滴定法绘制了HK-SEDDSs的伪三元相图,确定了处方中油相、表面活性剂和助表面活性剂的质量比范围,最终通过Box-Behnken实验设计得到HK-SEDDSs的最佳处方组成:capryol 90为油相,cremophor EL为表面活性剂,transcutol P为助表面活性剂,三者的质量比为10∶60∶30,用最优处方制备的HK-SEDDSs具有热力学稳定性好、自乳化速度快、形成的微乳透光率高、粒径小且分布均匀、药物溶出速度快的优点,为开展动物体内药动学评价奠定了药学基础。
药动学结果显示,口服HK-SEDDSs组大鼠的cmax和AUC(0→∞)均明显提高,分别是口服HK混悬剂组大鼠的1.6倍和1.6倍(P<0.05),这是由于HK-SEDDSs进入体内迅速形成粒径极小、比表面积极大的微乳,提高了药物的溶解度,促进了药物的吸收。
本研究制备的HK-SEDDSs虽然显著提高了HK的口服生物利用度,但是HK-SEDDSs是以液体状态存在,不利于药物的保存及患者的使用,后续会将HK-SEDDSs开发成易于储存和便于携带与使用的固体制剂。
