一种NHC-Cu(Ⅱ)配合物的合成及其对乙苯的催化氧化反应研究
2023-09-07杨喜庆程常君李成毛亚宁胡泉源
杨喜庆,程常君,李成,毛亚宁,胡泉源
(湖北大学化学化工学院, 湖北 武汉 430062)
0 引言
直接C—H键活化和官能化在惰性C—H化学的基础研究中具有重要意义,因其潜在的有机合成步骤经济性和绿色环保性[1-2],已成为有机化学中最活跃的研究领域[3-4].近年来,为了将简单烷烃和芳烃官能化有机化学家探索了各种行之有效的方法[5-6],在烷烃的硼化[7]和C—H键的氢原子转移过程[8]、烷烃氧化C(sp3)—H羰基化[9]等领域的研究都取得了重大进展.文献报道中,对乙基苯催化氧化常常使用无机复合金属组成的杂相催化剂[10-11],其产物苯乙酮的收率和选择性令人欣喜.
金属氮杂卡宾配合物在催化领域应用广泛[12-14],其作为有机氧化反应催化剂的研究常见诸报道[15-18],如NHC-Zn(Ⅱ)多相催化剂在温和条件下将芳醛氧化为相应的羧酸[15],NHC-Fe(Ⅱ)在甲苯中用叔丁基过氧化氢实现了醛与胺的氧化酰胺化反应[16],NHC-Pd(Ⅱ)催化无保护的1,2-和1,3-二醇选择性地氧化为羟基酮[17],用O/C/O型三齿配体的氮杂卡宾(NHC)铜Cu(Ⅱ)配合物催化乙基苯的氧化研究尚少见报道.
本研究报道新型NHC前体1,3-双(2-羟基-3,5-二叔丁基苄基)苯并咪唑氯化物及其Cu(Ⅱ)配合物的制备方法,以及作为催化剂应用于乙苯以及相应衍生物的氧化反应研究.
1 实验部分
1.1 试剂与仪器无水氯化铜、叔丁醇钾、六次甲基四胺(AR,国药集团有限公司)、硼氢化钠(AR,成都科龙化工试剂厂)、邻苯二胺、2,4-二叔丁基苯酚、原甲酸三乙酯、叔丁基过氧化氢、乙苯、1-甲氧基-4-丙基苯、4-乙基苯甲腈、1-氯-4-乙基苯、异丁基苯、4-正丙基苯乙酮(AR,伊诺凯有限公司).
400M核磁共振仪(Bruker公司,德国)、红外光谱仪(Thermo公司,中国)、紫外-可见/近红外分光光度计(日本岛津公司,日本)、X射线光电子能谱(Bruker公司,德国)、气相色谱仪(福立公司,中国).
1.2 实验过程
1.2.1 3,5-二叔丁基水杨醛的合成 该化合物参照文献[19]方法合成.1H NMR (400 MHz, CDCl3)δ:11.65 (s, 1H), 9.87 (s, 1H), 7.59 (d,J= 2.4 Hz, 1H), 7.35 (d,J= 2.5 Hz, 1H), 1.43 (s, 9H), 1.33 (s, 9H).13C{1H} NMR (100 MHz, CDCl3)δ:197.40, 159.14, 141.66, 35.05, 34.28.
1.2.2 N,N′-亚苯基-双(3,5-二叔丁基水杨醛亚胺)的合成 该化合物参照文献[20]方法合成.1H NMR (400 MHz, CDCl3)δ:13.54 (s, 2H), 8.66 (s, 2H), 7.44 (d,J= 2.4 Hz, 2H), 7.31 (dd,J= 5.9, 3.4 Hz, 2H), 7.24 (dd,J= 5.8, 2.5 Hz, 2H), 7.21 (d,J= 2.5 Hz, 2H), 1.44 (s, 18H), 1.32 (s, 18H).13C{1H} NMR (101 MHz, CDCl3)δ:164.75, 158.61, 142.79, 140.35, 137.22, 128.21, 127.34, 126.81, 119.83, 118.38, 35.14, 34.19, 31.50. 29.46.
1.2.3 N,N′-亚苯基-双(3,5-二叔丁基水杨醛胺)的合成 将N,N′-亚苯基-双(3,5-二叔丁基水杨醛亚胺2.70 g (5 mmol)、二氯甲烷和无水甲醇混合溶剂 (4∶1) 50 mL依次加入到100 mL的圆底单口烧瓶中,NaBH41.89 g (50 mmol)分批次加入,待溶液变为无色透明,停止反应.加水淬灭反应,乙酸乙酯萃取,有机相用无水硫酸钠干燥,过滤,旋干得到浅白色固体,65 ℃真空干燥,产物2.12 g,收率77.94%.1H NMR (400 MHz, CDCl3)δ:7.99 (s, 2H), 7.28 (s, 2H), 7.05 (s, 2H), 6.99 (m, 4H), 4.37 (s, 4H), 3.61 (s, 2H), 1.39 (s, 18H), 1.30 (s, 18H).13C{1H} NMR (101 MHz, CDCl3)δ:153.19, 141.92, 136.77, 136.32, 124.10, 123.92, 122.18, 121.76, 114.47, 48.88, 34.98, 34.30, 31.69, 29.79.
1.2.4 1,3-双(2-羟基-3,5-二叔丁基苄基)苯并咪唑氯化铵(L)的合成 将N,N′-亚苯基-双(3,5-二叔丁基水杨醛胺)2.72 g (5 mmol)、原甲酸三乙酯50 mL依次加入到100 mL的圆底单口烧瓶中,再加入浓盐酸2 mL(12 mol/L, 0.167 mol),均匀搅拌,逐渐升温到120 ℃反应,12 h后停止反应.冷却至室温,有固体析出,过滤,滤饼用乙酸乙酯洗涤,得到白色固体,65 ℃真空干燥,产物2.25 g,收率76.27%.1H NMR (400 MHz, DMSO-d6)δ:9.24 (s, 1H), 8.81 (s, 2H), 8.03 (dd,J= 6.3, 3.1 Hz, 2H), 7.67 (dd,J= 6.3, 3.1 Hz, 2H), 7.23 (m, 4H), 5.77 (s, 4H), 1.34 (s, 18H), 1.19 (s, 18H).13C{1H} NMR (101 MHz, DMSO-d6)δ:152.01, 142.92, 141.56, 139.30, 131.82, 127.05, 125.76, 124.77, 122.30, 114.49, 47.57, 35.22, 34.43, 31.76, 30.26. MS: C37H51ClN2O2: 590.363 3 (Theo.), 555.420 97(M—Cl, Found).
1.2.5LCu(Ⅱ)的合成 称取配体L1.77 g (3 mmol)、叔丁醇钾1.01 g (9 mmol)依次加入到50 mL Schlenk反应瓶,在氮气保护下,注入20 mL无水THF,反应1 h后,缓慢加入到无水氯化铜(0.54 g, 4 mmol)的丙酮溶液中,继续反应24 h.将反应液减压浓缩,加入足量的水超声分散,固体过滤,滤饼分别使用纯水和少量甲醇洗涤,65 ℃真空干燥12 h,得到黄绿色固体产物1.60 g,收率77.66%. MS: C40H54N2O3Cu:673.342 5(Theo.), 673.456 85 (Found ),如图1所示.
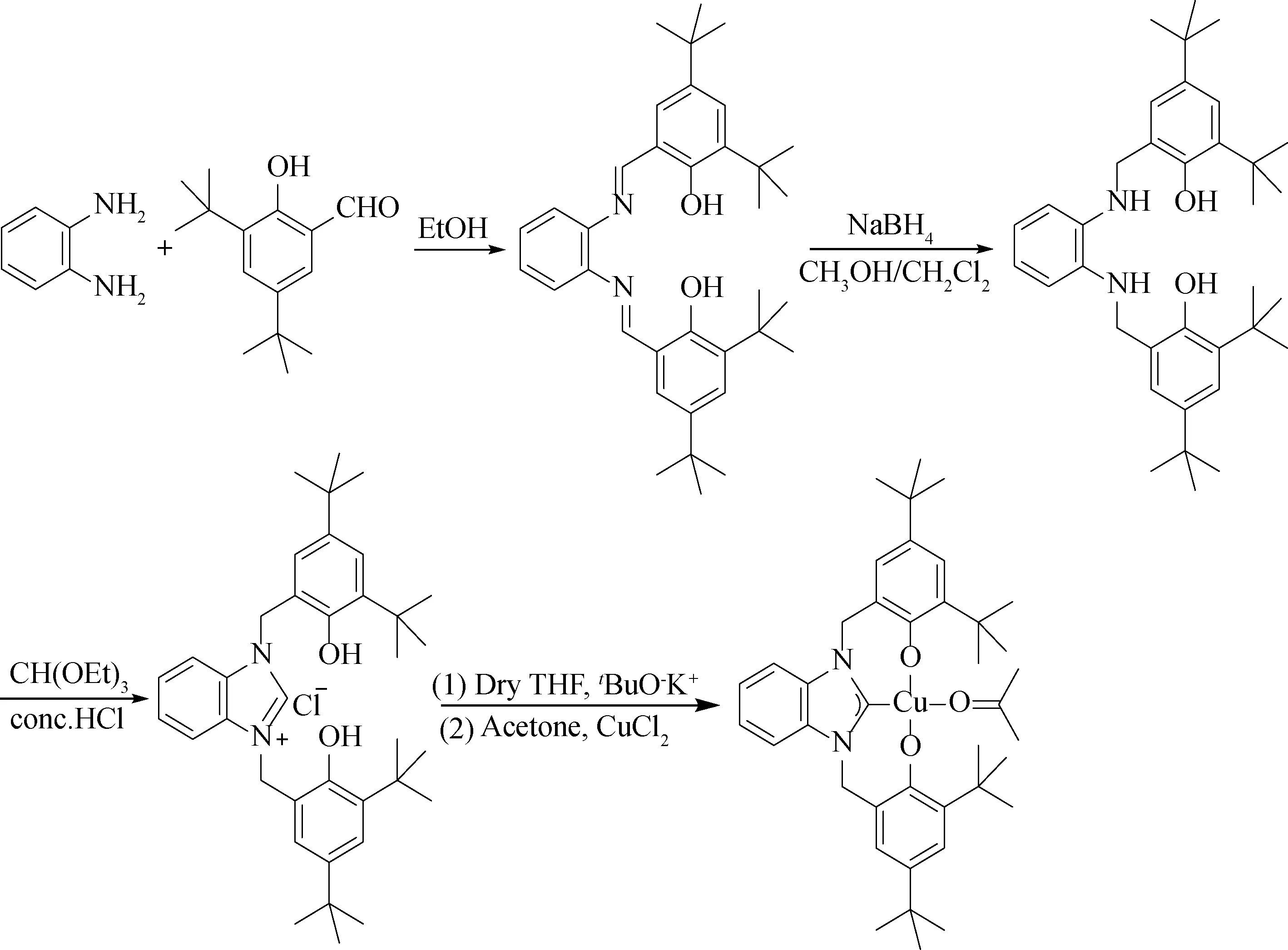
图1 LCu(Ⅱ)的合成路线
1.2.6 催化氧化反应 称取5 mmol底物放入25 mL的Schlenk管中,加入10 mL乙腈和1%(物质的量)LCu(Ⅱ)催化剂,最后加入一定量的氧化剂(TBHP),一定温度下搅拌,反应12~24 h.中途取样,利用气相色谱仪检测反应结果(三甲苯作为内标),每个样品重复两次,计算原料的转化率和产物选择性.GC分离条件:采用HP-5石英毛细管柱(30 m×0.32 mm×0.25 μm)、柱温80 ℃、进样器250 ℃、检测器250 ℃,使用程序升温80 ℃保留3 min,然后10 ℃/min升温到250 ℃保留3 min.
2 结果与讨论
2.1 催化剂的结构表征
2.1.1 UV-vis表征 图2显示的是配体L和配合物的紫外-可见吸收光谱图.图谱中配体L的最大吸收峰230 nm和270 nm分别π→π*跃迁和n→π*跃迁信号,在配体L与Cu2+配位后,230 nm吸收峰为π→π*跃迁信号,n→π*跃迁信号减弱接近消失,440 nm吸收峰明显来自于金属离子和配体之间的电荷转移效应(MLCT).
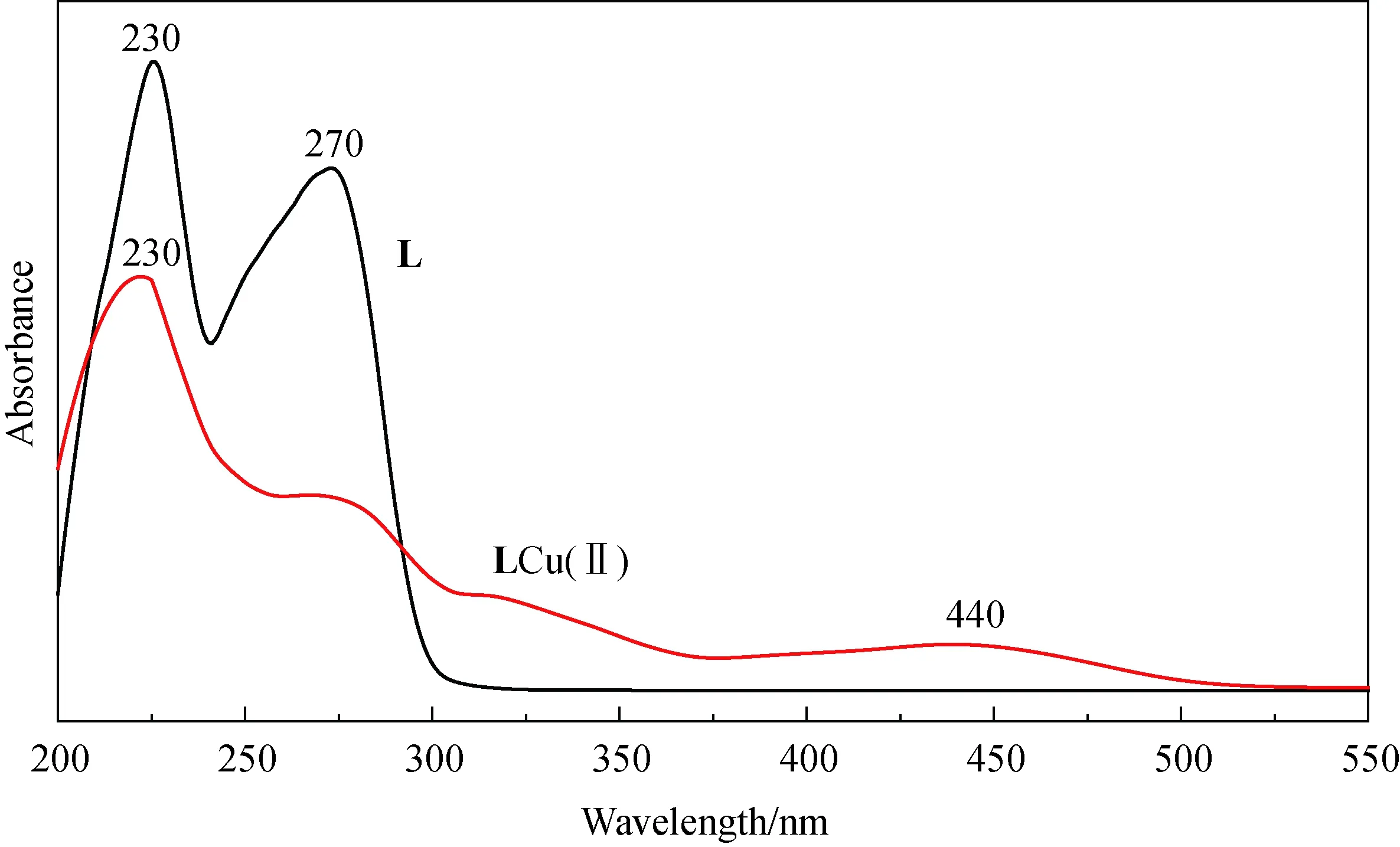
图2 配体L和LCu(Ⅱ)UV-vis光谱
2.1.2 红外图谱表征 图3显示的是配体L和配合物的红外图谱.从配体L的红外图谱中可知,3 680 cm-1处为酚O—H的伸缩振动吸收峰,1 210 cm-1处为酚的C—O的伸缩振动吸收峰,1 300 cm-1处为酚O—H的面内弯曲振动吸收峰,3 160 cm-1处为季铵盐C—N的伸缩振动吸收峰,表现为宽的吸收,与Cu2+配位后,酚O—H和季铵盐C—N的吸收峰消失相比于配体L,图谱中苯环骨架峰1 480 cm-1、1 610 cm-1吸收峰增强.
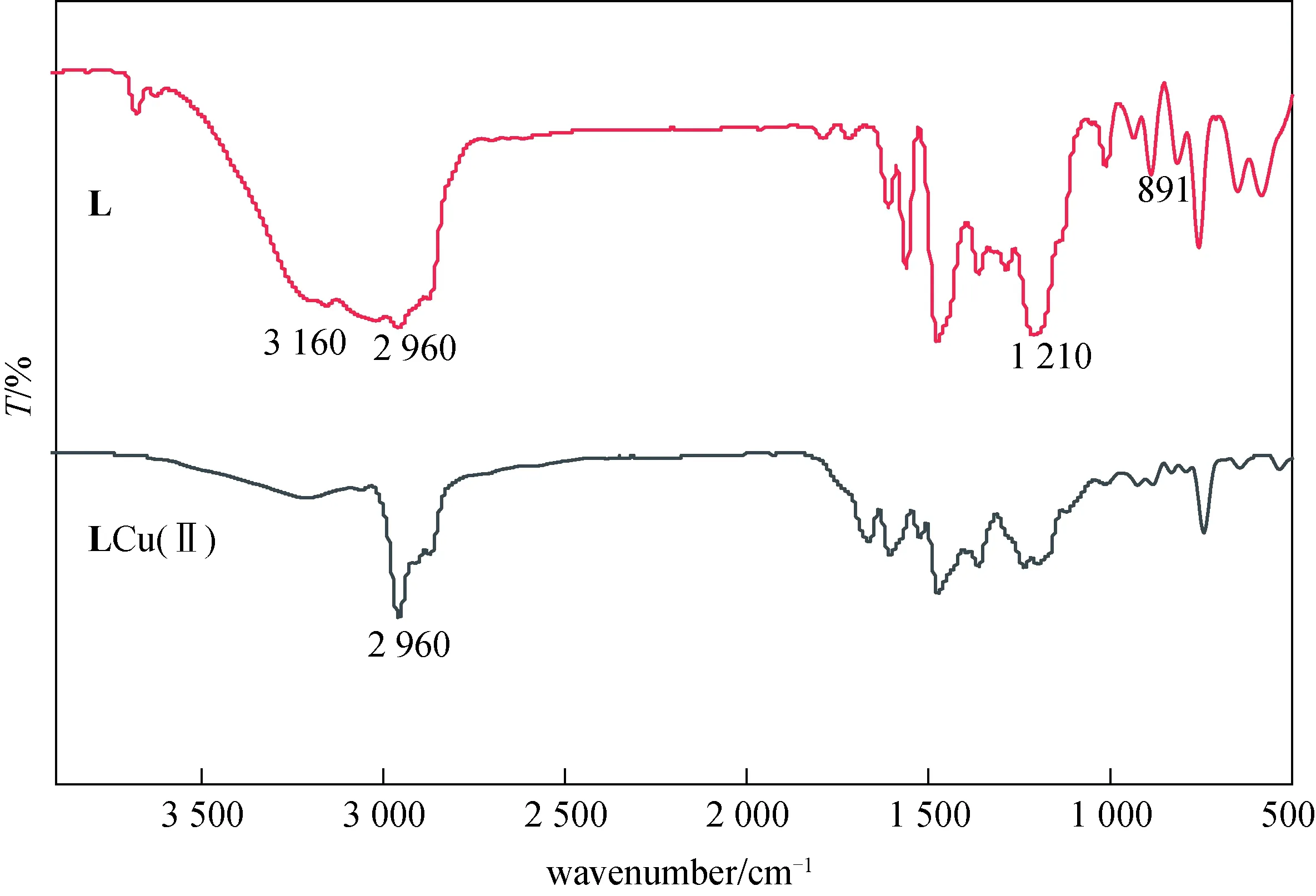
图3 配体L和LCu(Ⅱ)红外图谱
2.1.3 XPS表征 图4显示的是配合物的X射线光电子能谱,其中C,80.22%(质量百分数),N,6.49%(质量百分比), O,11.26%(质量百分比),Cu,2.03%(质量百分比).C1s、N1s、O1s、Cu2p的结合能分别在284 eV、400 eV、531 eV、933 eV,没有出现Cl元素的吸收信号,说明形成配合物时Cu—Cl完全被Cu—O取代.图5显示的是配合物的X射线光电子能谱Cu精细谱,Cu 2p3、Cu 2p1的结合能分别在932.6 eV、952.6 eV,表明了Cu2+的存在.
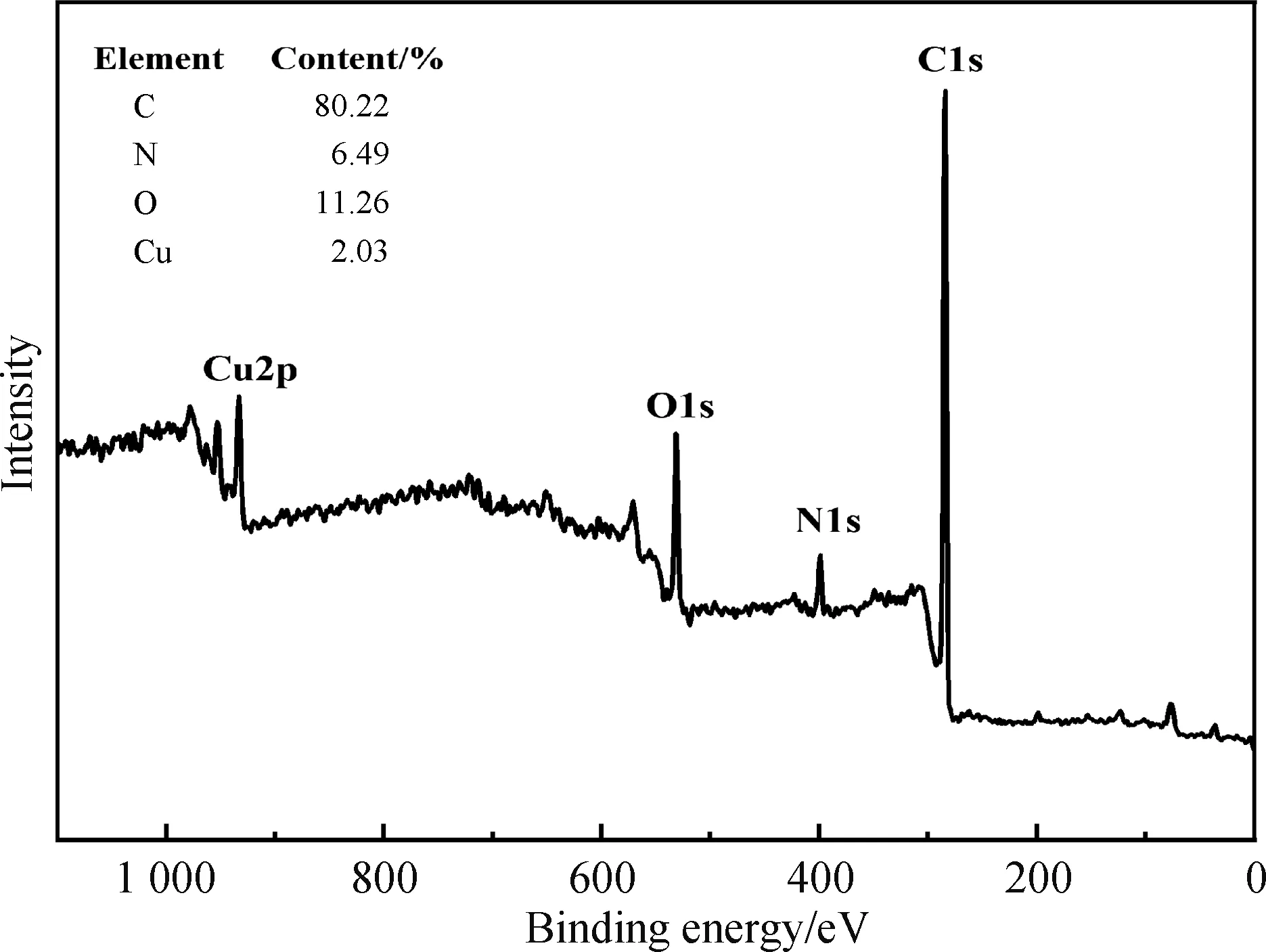
图4 LCu(Ⅱ)的X射线光电子能谱
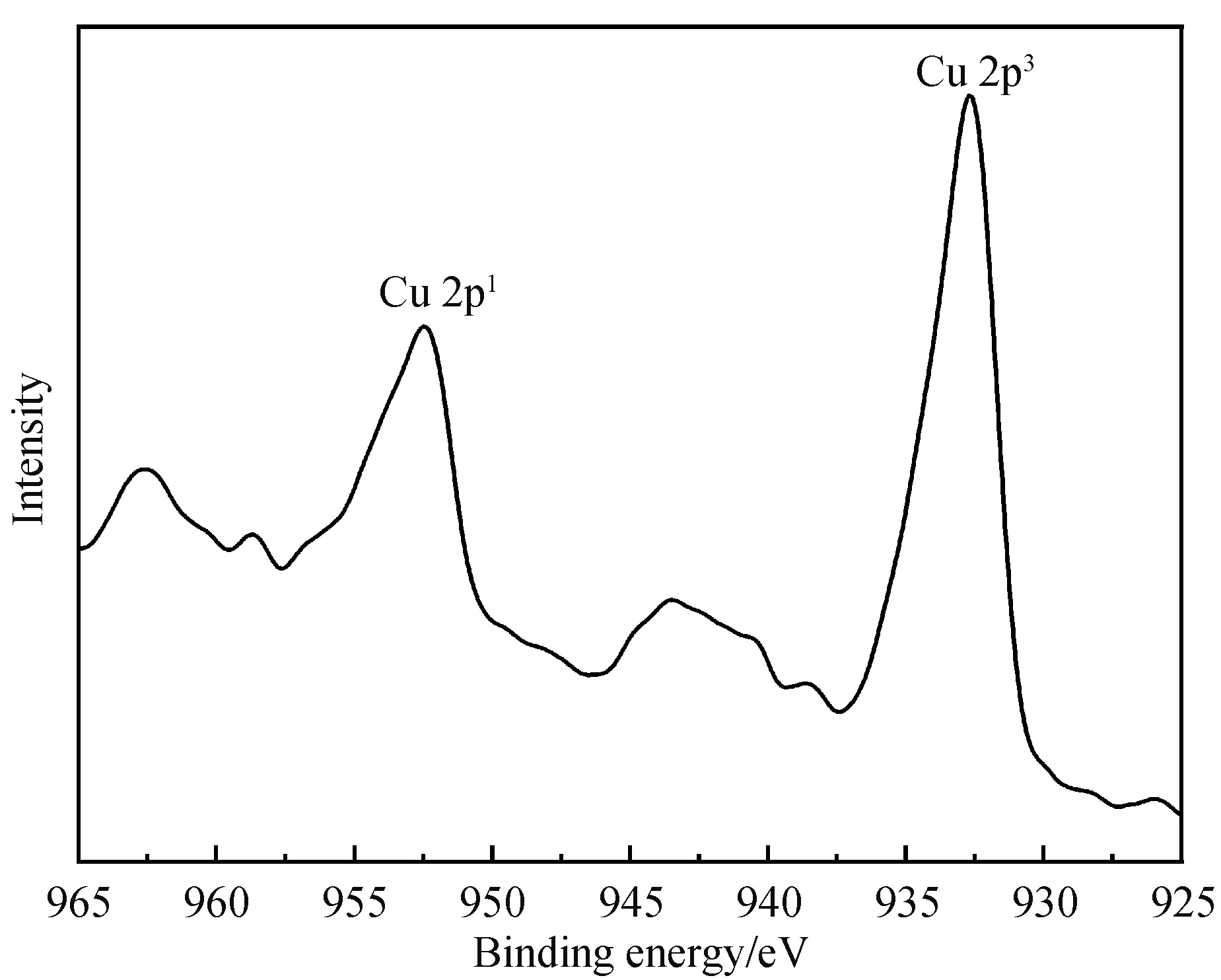
图5 LCu(Ⅱ)的X射线光电子能谱Cu精细谱
2.2 催化性能试验以乙苯为底物,以TBHP为氧化剂,LCu(Ⅱ)为催化剂,该反应的产物为苯乙醇和苯乙酮,无其他杂质.为了探究催化氧化的最优反应条件,分别对反应物投料比、反应温度、反应溶剂和催化剂投料量进行了系列研究,实验见表1至表4.
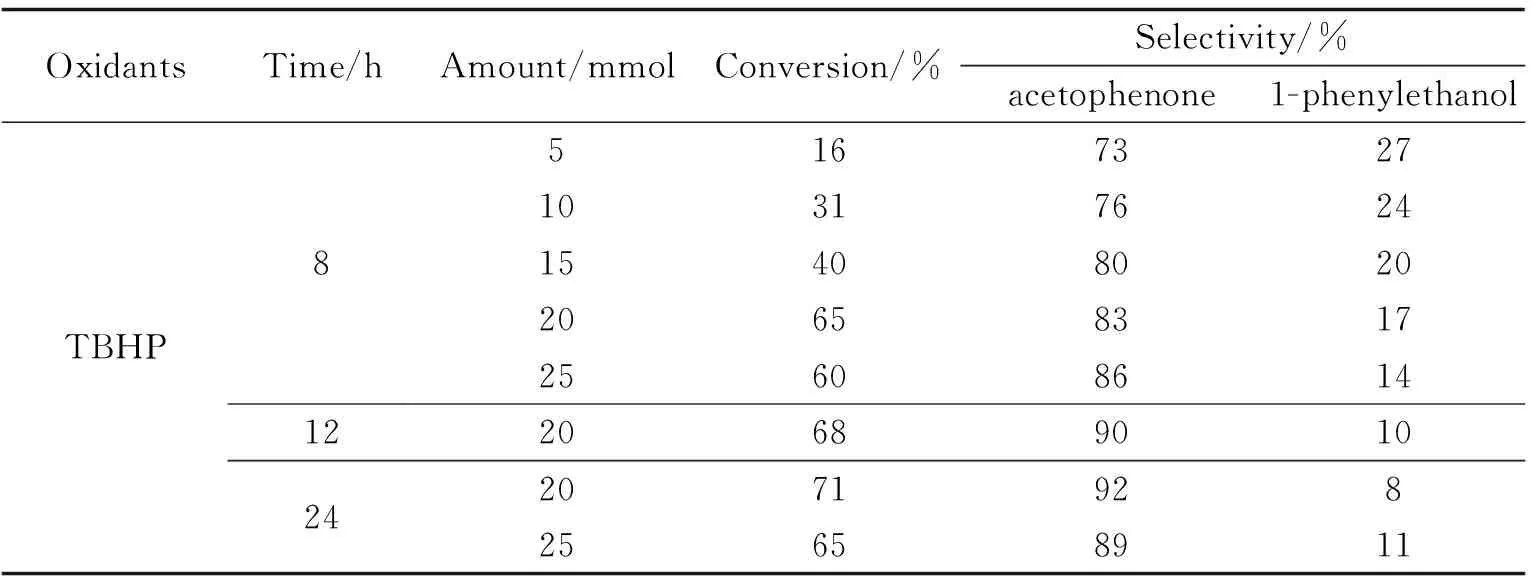
表1 乙苯-TBHP比例对乙苯氧化的影响
探究乙苯-TBHP比例对乙苯的影响,表1中可以发现当乙苯-TBHP的摩尔投料比为1∶4时,效果最佳.反应8 h后乙苯的转化率即可到65%,其后增加缓慢,12 h为68%,24 h为71%.反应中间体为苯乙醇,可被继续氧化为苯乙酮,故苯乙酮的选择性随时间逐步提高,12 h即可到达90%,
但当乙苯-TBHP的比例增加到1∶5时,反应效果反而不及比例为1∶4的情况,这是因为TBHP通过与催化剂中过渡金属发生协同作用而激活,tBuOOH可以利用其羟基(-OH)与铜(II)空位配合形成中间产物{tBuO-O-Cu},有利于乙苯氧化生成所需产物[21],TBHP浓度过高反而不利于产物从催化中心原子上离去.
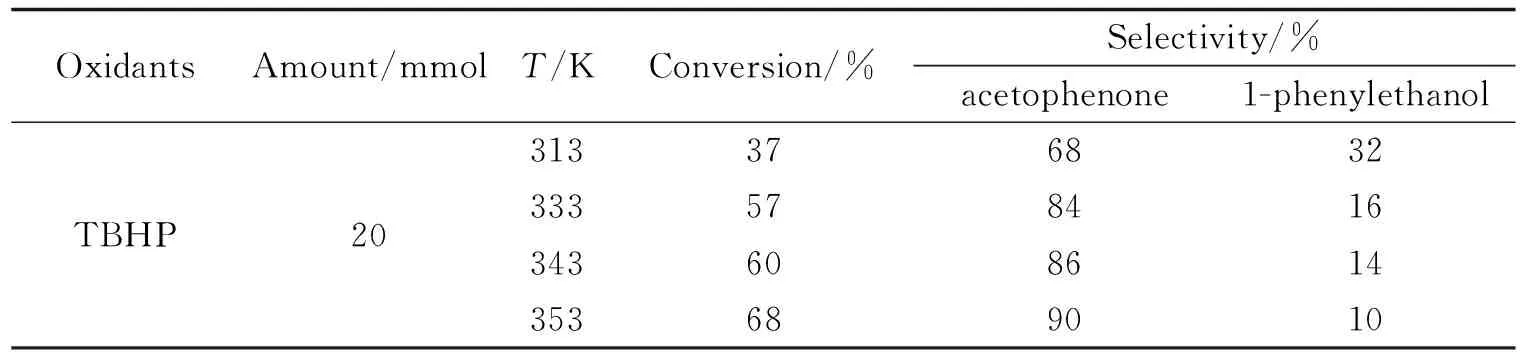
表2 温度对乙苯氧化的影响
探究温度对乙苯氧化的影响,表2中可以发现当在313、333、343和353 K的测试温度下,随着温度的升高,乙苯的转化率也在升高,这说明氧化需要较高的反应温度.当温度为353 K时,12 h后乙苯的转化率达到68%,苯乙酮的选择性达到90%.

表3 溶剂对乙苯氧化的影响
探究溶剂对乙苯氧化的影响.表3中可以发现采用不同极性的溶剂被用作反应溶剂时,乙腈的效果最好.在353 K时,不同溶剂对乙苯催化氧化活性排序为CH3CN>DMF>DMSO>CH2Cl2,溶剂条件下转化率的降低归因于溶剂分子对催化剂活性位点的阻断[22].

表4 催化剂投料对乙苯氧化的影响
探究催化剂投料对乙苯氧化的影响.表4中可以发现当催化剂投料从0.5%(mol)增加到1%(mol)时,乙苯的转化率从56%上升到68%,当继续增加催化剂投料到2%(mol)时,乙苯的转化率反而下降.可能的原因是过量的催化剂会分解活性的过氧化物中间体,使其失活,影响催化效率.为了验证催化剂的存在与否对反应的影响,进行了空白对照实验.表明乙苯的转化率只有20%,不使用催化剂是一个原料的自氧化过程.
2.3 机理探索到目前为止,大多数的文献报道乙苯的氧化过程是自由基反应过程[23-24].根据这个催化实验结果,推测该反应机理如图6所示.
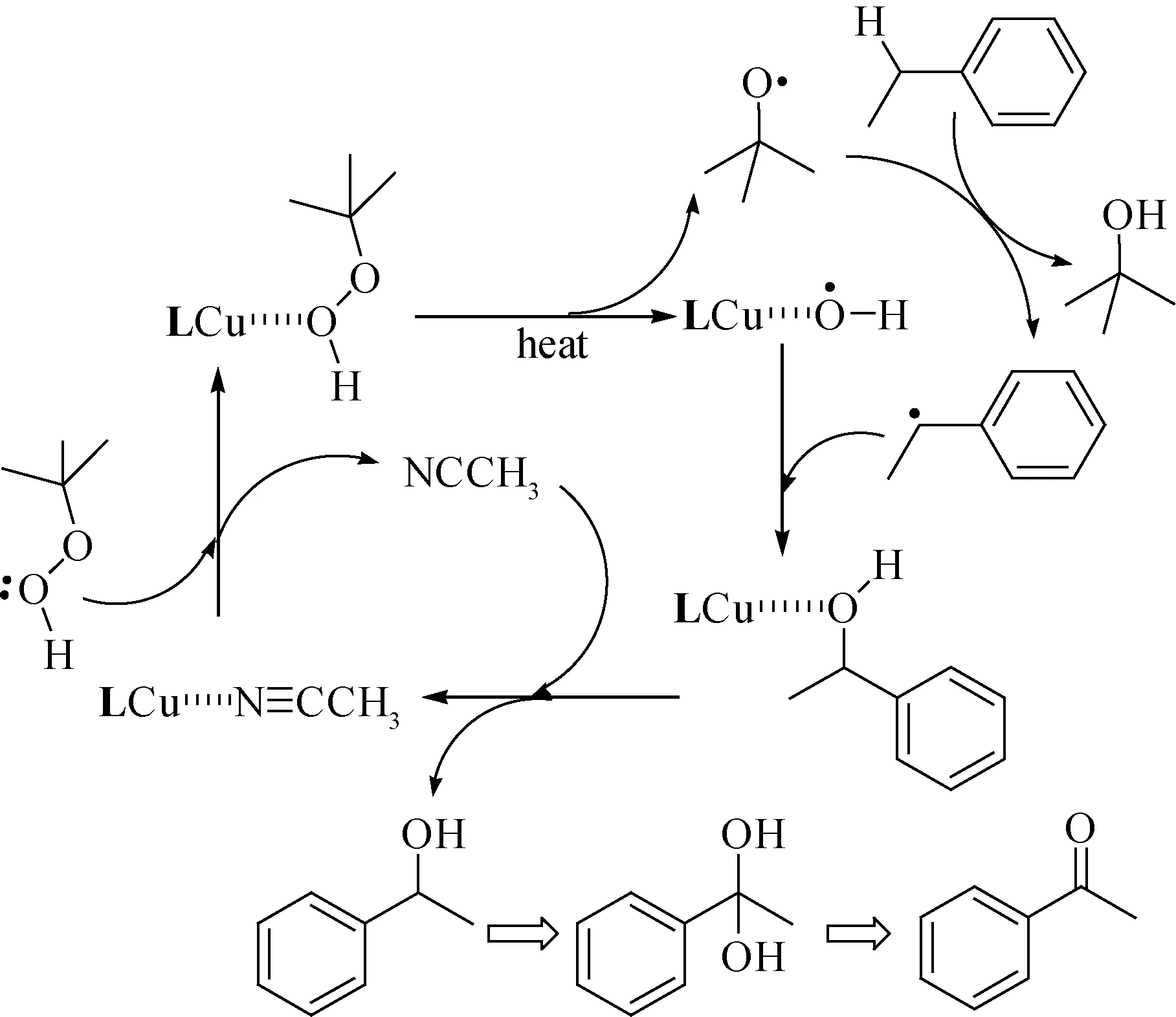
图6 乙苯氧化推测的反应机理
首先TBHP对催化剂LCu(Ⅱ)上弱配位的乙腈分子交换,外界提供能量激活TBHP分子中的O—O键的均裂,产生叔丁氧自由基(tBuO·)和活性催化中间体{[LCu-OH]·}.tBuO·夺取乙苯的Cα—H,生成α-苯乙基自由基,该自由基可以结合活性催化中间体{[LCu-OH]·}生成中间产物,最后α-苯乙醇脱离催化活性中心,其空位被乙腈分子占据,完成催化循环.
α-苯乙醇的剩余Cα—H继续被tBuO·进攻,发生上述类似催化过程,在Cα上形成二羟基取代产物,该产物不稳定,易发生二羟基重排,形成α-乙苯过氧化氢中间体,最后脱水产生苯乙酮.
2.4 乙苯衍生物催化性能实验为了拓展底物的应用范围,选择了部分乙苯衍生物进行了氧化实验探究,实验结果见表5.研究发现当烷基对位连有吸电子基时,底物的转化率更高,对应酮的选择性也更高,这是因为受诱导效应的影响,烷基α-C电子云密度降低,Cα—H共价键强度减弱,α-H更容易被自由基进攻而离去.烷基碳链增长时,底物的转化率较低,可能是因为空间效应引起的.

表5 底物催化氧化结果
3 结论
本工作报道了一种新型氮杂卡宾配合物NHC-Cu(Ⅱ)的合成和表征,探讨了该化合物作为催化剂对乙苯及其衍生物的催化氧化活性.研究结果表明,当TBHP用作氧化剂时该催化剂在353 K、乙腈作溶剂下表现出了较好的催化氧化活性,催化剂的使用量仅为1%(mol),乙苯在反应12 h后的转化率达到68%,其产物苯乙酮的选择性达到90%.对类似结构衍生物的拓展氧化实验结果显示,烷基对位有吸电子取代基时,相同条件下底物的转化率和相应酮的选择性更高,研究结论对于有机化合物C—H氧化及催化剂开发领域具有积极的意义.
