基于UPLC特征图谱和一测多评法的马鞭草质量评价及其标准汤剂量值传递规律研究
2023-09-02杨晓东罗宇琴吴文平潘礼业刘晓琳邢菊玲李国卫
杨晓东,罗宇琴,吴文平,潘礼业,刘晓琳,庞 伟,邢菊玲,李国卫
广东一方制药有限公司 广东省中药配方颗粒企业重点实验室,佛山 528244
马鞭草为马鞭草科植物马鞭草VerbenaofficinatisL.的干燥地上部分[1],始载于《名医别录》,列为下品。马鞭草的化学成分主要为环烯醚萜苷类[2]、黄酮类[3]、甾醇类、三萜类、多酚类和挥发油[4]、鞣质、β-胡萝卜素,咖啡酸,糖类等。主要活性成分为环烯醚萜苷类成分[5]。其性凉,味苦,归肝、脾经。具有活血散瘀、解毒、利水、退黄、截疟的功效。临床用于癥瘕积聚,痛经经闭,喉痹,痈肿,水肿,黄疸,疟疾。马鞭草资源丰富[6],其中环烯醚萜苷类[7]成分具有抗氧化、保肝、保护神经[8]、保护心脏及脑缺血[9]、抗炎[10]等多种作用。中药汤剂(标准汤剂)是中医临床最常用的中药复方制剂,也是中医历史上应用最久和最广的制剂。无论是中药传统饮片,还是破壁饮片、中药配方颗粒等中药新型饮片,均是采用水煎服或者水冲服,因此,将标准汤剂研究作为其他中药制剂研究的首选对照具有十分重要的意义[11]。本研究通过阐明饮片到标准汤剂的量值传递规律,为马鞭草中药制剂工艺研究和质量评价奠定基础。
本研究以马鞭草饮片为研究对象,建立其UPLC特征图谱,结合化学模式分析图谱信息并采用超高效液相色谱法联合一测多评法同时测定马鞭草中毛蕊花糖苷、5-羟基马鞭草苷、马鞭草苷含量的方法,与外标法测定结果进行比较。并测定其标准汤剂出膏率、毛蕊花糖苷、5-羟基马鞭草苷及马鞭草苷含量,计算转移率,以此为基础进行马鞭草饮片到标准汤剂的量值传递研究,说明各项质量指标设定的合理性。
1 材料
1.1 仪器
Waters H-Class型超高效液相色谱仪(沃特世公司);Thermo Vanquish型超高效液相色谱仪(赛默飞科技有限公司);ME204E型万分之一天平(梅特勒-托利多公司)。
1.2 试剂
甲醇(西陇科学股份有限公司)为分析纯;液相用磷酸(天津市科密欧化学试剂有限公司);乙腈(默克股份有限公司)为色谱纯;水为超纯水(实验室自制)。
1.3 试药
马鞭草苷(成都瑞芬思生物科技有限公司,批号:M-042-170613,含量:98.0%);5-羟基马鞭草苷(维克奇生物科技有限公司,批号:wkq18110104,含量:98.0%);毛蕊花糖苷(中国食品药品检定研究院,批号:111530-201713,含量:92.5%)。本次实验共收集18批马鞭草药材,产地信息(见表1),经广东一方制药有限公司孙冬梅教授鉴定,均为马鞭草科植物马鞭草VerbenaofficinatisL.的干燥地上部分,并经广东一方制药有限公司质量中心鉴定合格,均符合2020版《中华人民共和国药典》(后文简称《中国药典》)马鞭草项下的各项规定,并按2020版《中国药典》马鞭草饮片项下炮制成饮片,具体炮制方法为:取马鞭草药材,挑去非药用部位和杂质,洗净,稍润,切成5~15 mm的段,60 ℃烘干。

表1 样品信息Table 1 Sample information
2 方法与结果
2.1 色谱条件
采用Waters HSS T3(100 mm×2.1 mm,1.8 μm)色谱柱;流动相为乙腈(A)-0.05%磷酸水溶液(B),梯度洗脱(0~2 min,10% A;2~14 min,10%→18% A;14~16 min,18% A;16~25 min,18%→25% A;25~28 min,25%→95% A);流速为0.3 mL/min;柱温为35 ℃;检测波长为230 nm;进样量为1 μL。
2.2 对照品及供试品溶液制备
2.2.1 对照品溶液配制
取马鞭草苷对照品、5-羟基马鞭草苷对照品、毛蕊花糖苷对照品适量,精密称定,置量瓶中,加甲醇制成每1 mL含马鞭草苷30 μg,5-羟基马鞭草苷76 μg,毛蕊花糖苷22 μg的混合溶液,即得。
2.2.2 饮片供试品溶液制备
取马鞭草饮片粉末(过三号筛)约1 g,精密称定,置锥形瓶中,精密加入80%甲醇50 mL,称定重量,加热回流120 min,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2.2.3 标准汤剂供试品溶液制备
取马鞭草标准汤剂约0.1 g,精密称定,置锥形瓶中,精密加入80%甲醇20 mL,称定重量,超声处理(功率300 W,频率40 kHz)30 min,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2.3 马鞭草UPLC特征图谱建立
2.3.1 特征图谱方法学考察
2.3.1.1 精密度考察
取马鞭草饮片粉末(过三号筛,编号:S1),按“2.2.2”项下方法制备供试品溶液,按“2.1”项下色谱条件连续进样6次,以毛蕊花糖苷峰为参照峰,各特征峰相对保留时间和相对峰面积RSD均小于2.0%,表明仪器精密度良好。
2.3.1.2 重复性考察
取马鞭草饮片粉末(过三号筛,编号:S1),按“2.2.2”项下方法平行制备6份供试品溶液,按“2.1”项下色谱条件进样测定,以毛蕊花糖苷峰为参照峰,各特征峰相对保留时间和相对峰面积RSD均小于2.5%,表明样品制备方法重复性良好。
2.3.1.3 稳定性考察
取马鞭草饮片粉末(过三号筛,编号:S1),按“2.2.2”项下方法制备供试品溶液,按“2.1”项下色谱条件分别与0、2、4、6、8、12 h进样测定。以毛蕊花糖苷峰为参照峰,各特征峰相对保留时间和相对峰面积RSD均小于2.1%,表明样品在12 h内稳定。
2.3.2 特征图谱的建立及相似度评价
取18批马鞭草饮片按“2.2.2”项下方法制备供试品溶液,按“2.1”项下色谱条件进样检测。将色谱图导入《中药色谱指纹图谱相似度评价系统》(2012年版),设定S1为参照图谱,自动匹配后多点校正,进行全谱峰匹配,得到18批样品叠加图,共确认5个特征峰(见图1)。采用《中药色谱指纹图谱相似度评价系统》(2012年版)计算18批马鞭草饮片特征图谱与对照指纹图谱的相似度。结果显示,18批样品的相似度均不低于0.83(见表2)。
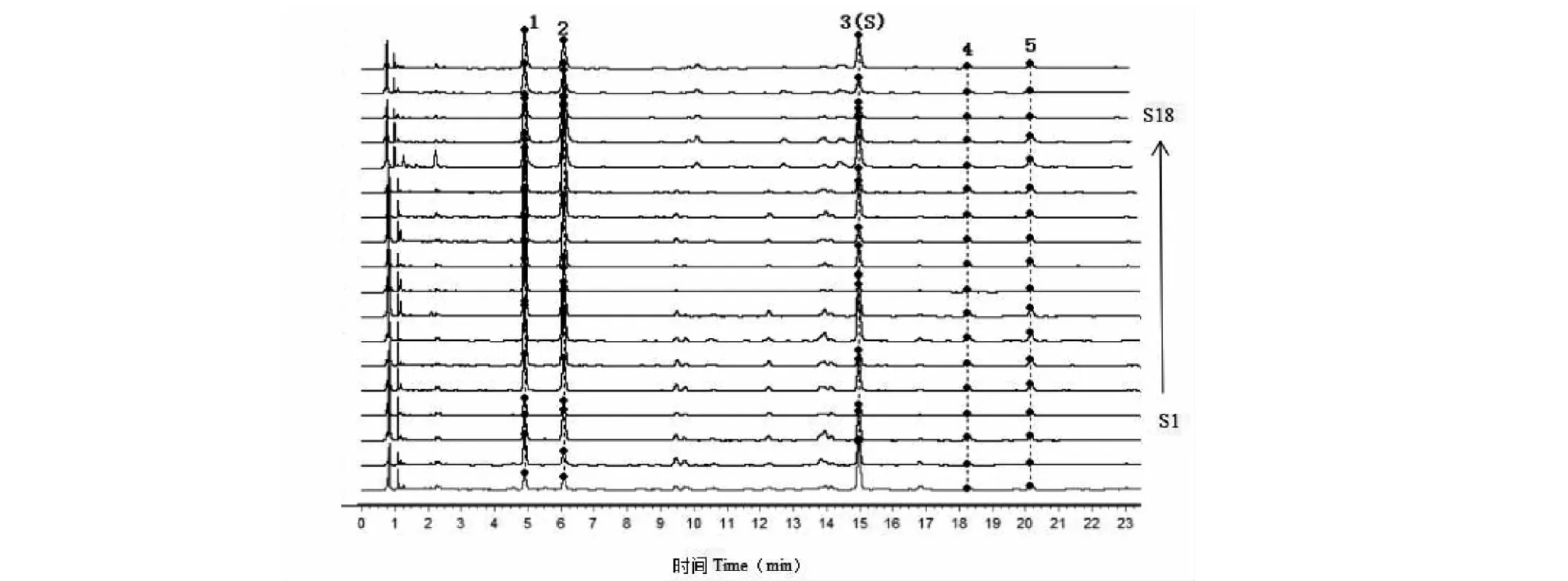
图1 18批马鞭草特征图谱Fig.1 Characteristic chromatogram of 18 batches of Verbenae Herba

表2 相似度结果Table 2 Results of the similarity
2.3.3 共有峰指认
为考察特征图谱是否具有代表性及能否表征待测成分的专属性,通过与对照品对照,指认其中3个特征峰,峰1为5-羟基马鞭草苷、峰2为马鞭草苷、峰3为毛蕊花糖苷(见图2)。
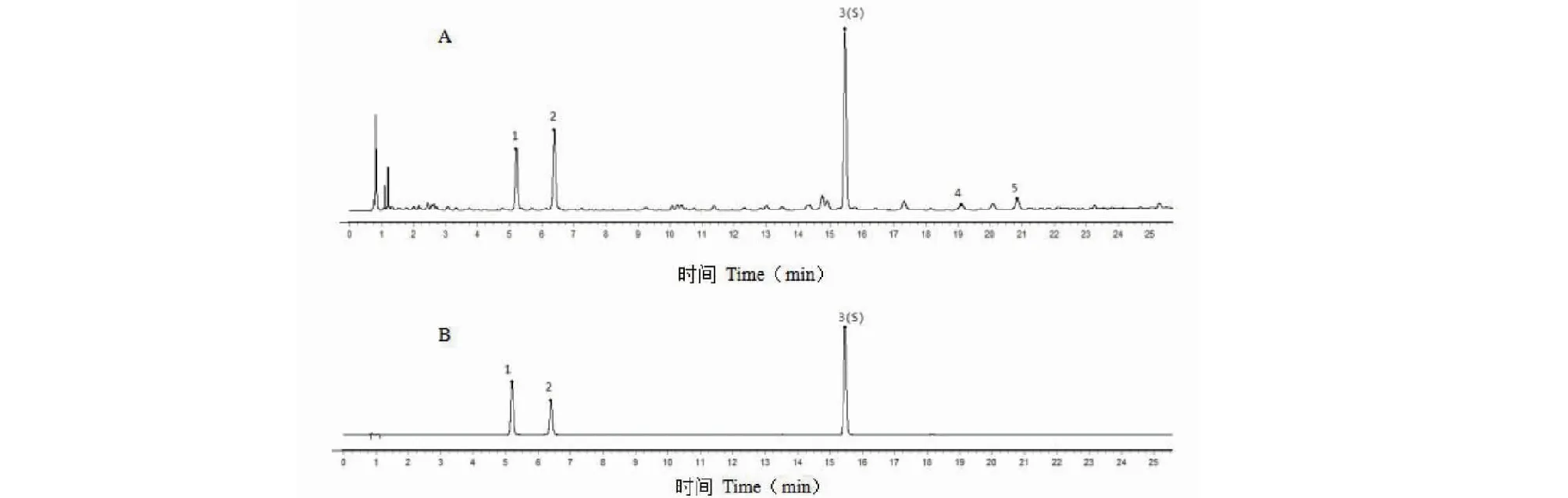
图2 特征图谱共有峰的指认Fig.2 Identification of common peaks in characteristic chromatogram注:A:供试品;B:混合对照品。1:5-羟基马鞭草苷;2:马鞭草苷;3:毛蕊花糖苷。Note:A:Sample;B:Mixed reference substance.1:Hastatoside;2:Cornin;3:Verbascoside.
2.3.4 主成分分析
以18批马鞭草饮片特征图谱中的5个共有峰峰面积为数据,用SPSS 20.0软件对其进行主成分分析,得出相关矩阵的特征值及其方差(见表3),碎石图(见图3),由表3可知,根据特征值大于1,得到两个主成分,对总方差的累计贡献度达80.766%,具有很好的代表性,可代表马鞭草特征图谱共有峰的大部分信息。成分矩阵(见表4),由表4可知,第1主成分主要反应了峰3(毛蕊花糖苷)和峰5的信息,第二主成分反映了峰1(5-羟基马鞭草苷)、峰2(马鞭草苷)的信息。

图3 主成分分析碎石图Fig.3 Scree plot of principal component analysis

表3 特征值及累积方差贡献率Table 3 Eigenvalue and cumulative variance contribution rate
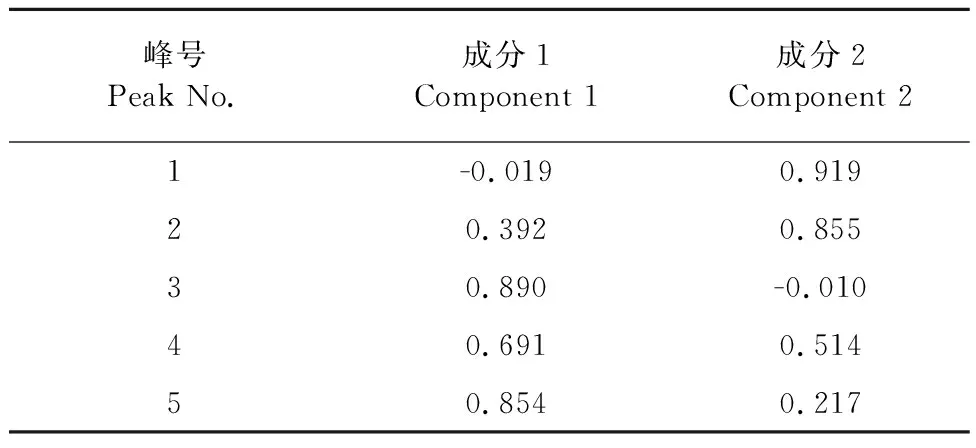
表4 主成分分析矩阵Table 4 Matrix of principal component analysis
2.4 一测多评法建立
根据主成分分析结果,以峰3(毛蕊花糖苷)、峰1(5-羟基马鞭草苷)、峰2(马鞭草苷)对马鞭草饮片整体质量影响较大。三者光谱特性基本一致且响应值比例接近,因此,以毛蕊花糖苷为内参物,马鞭草苷和5-羟基马鞭草苷为待测成分,建立上述三种成分的一测多评定量表征方法。
2.4.1 一测多评法方法学考察
2.4.1.1 精密度考察
取马鞭草饮片粉末(过三号筛,编号:S1),按照“2.2.2”项下方法制备供试品溶液,按“2.1”项下色谱条件连续进样6次、5-羟基马鞭草苷、马鞭草苷、毛蕊花糖苷峰面积RSD分别为0.27%、0.56%、0.70%,表明仪器精密度良好。
2.4.1.2 重复性考察
取马鞭草饮片粉末(过三号筛,编号:S1),按照“2.2.2”项下方法平行制备6份供试品溶液,按“2.1”项下色谱条件进样测定,计算马鞭草中5-羟基马鞭草苷、马鞭草苷、毛蕊花糖苷的含量,3种成分含量RSD均小于3%,表明样品制备方法重复性良好。
2.4.1.3 稳定性考察
取马鞭草饮片粉末(过三号筛,编号:S1),按照“2.2.2”项下方法制备供试品溶液,按“2.1”项下色谱条件分别在0、2、4、6、8、12 h进样测定。5-羟基马鞭草苷、马鞭草苷、毛蕊花糖苷峰面积RSD均小于1%。表明供试品溶液在12 h内稳定。
2.4.1.4 线性关系考察
取马鞭草苷对照品、5-羟基马鞭草苷对照品、毛蕊花糖苷对照品适量,精密称定,置量瓶中,加甲醇制成5-羟基马鞭草苷、马鞭草苷、毛蕊花糖苷浓度分别为757.305、463.932、1038.220 μg/mL的混合对照品储备溶液。
精密吸取上述混合储备液1 mL至2、5、10、20、50、100 mL量瓶,加甲醇定容至刻度,同上述对照品储备液分别按照“2.1”项下色谱条件依次进样测定,记录色谱峰峰面积。以峰面积为纵坐标(y),对照品浓度为横坐标(x),并绘制标准曲线(见表5)。结果表明,3种成分在线性范围内线性关系良好。

表5 回归方程及相关系数Table 5 Linear equation and correlation coefficient
2.4.1.5 加样回收率考察
取已测定含量马鞭草(S1)饮片粉末约0.5 g,精密称定,平行3组,每组3份,分别按1∶0.5、1∶1、1∶1.5加入5-羟基马鞭草苷、马鞭草苷、毛蕊花糖苷对照品;按照“2.2.2”项下方法制备供试品溶液,按“2.1”项下色谱条件进样测定,计算加样回收率,3种成分平均加样回收率在94.4%~102.04%范围内。RSD均小于3.0%,均符合2020年版《中国药典》四部通则9101的规定。表明该测定方法准确度良好。
2.4.2 一测多评法建立
2.4.2.1 相对校正因子计算
因毛蕊花糖苷含量相对较高,且其对照品较易获得,本研究以毛蕊花糖苷为内参物(s)与其他待测成分为(i)的相对校正因子的计算公示为
(1)
其中,RCF代表相对校正因子、s代表内参物、i代表其他待测成分、A为峰面积、C为相应对照品浓度。
2.4.2.2 耐用性考察
利用相对保留时间法,以毛蕊花糖苷峰为参照峰,计算5-羟基马鞭草苷、马鞭草苷的相对保留时间ris=tRi/tRs(式中,tRs为参照峰的保留时间,tRi为待测成分的保留时间)。对各色谱峰进行定位。精密吸取“2.4.1.1”项下样品溶液,分别考察不同柱温(33、35、37 ℃)、流速(0.28、0.30、0.32 mL/min)、色谱柱(YMC Triart、Waters HSS T3)、色谱仪(Waters H-Class、Thermo Vanquish)对相对保留时间的影响(见表6),RSD小于2%。

表6 耐用性考察(相对保留时间)Table 6 Durability investigation(relative retention time)
2.4.2.3 相对校正因子重现性考察
分别对柱温(33、35、37 ℃)、流速(0.28、0.30、0.32 mL/min)、色谱柱(YMC Triart、Waters HSS T3)、色谱仪(Waters H-Class、Thermo Vanquish)进行耐用性考察,取“2.4.1.1”项下样品,按“2.1”项下色谱条件进样测定,计算5-羟基马鞭草苷、马鞭草苷的相对校正因子(见表7),RSD小于2%。

表7 不同条件对相对校正因子的影响Table 7 Effects of different conditions on relative correction factors (RCF)
2.4.2.4 样品测定
取18批马鞭草饮片样品,按“2.2.2”项下方法制备供试品溶液,平行两份,按照“2.1”项下色谱条件进样测定,记录峰面积。以毛蕊花糖苷为参照物,按照一测多评法计算5-羟基马鞭草苷、马鞭草苷的含量。并与外标法测定结果(毛蕊花糖苷除外)进行比较,计算相对误差RE=(WQAMS-WESM)/WESM×100%(式中,除WESM为外标法测得的成分含量,WQAMS为一测多评法测得的成分含量)[12]。结果显示,18批样品两种方法测定结果的RE%均小于3%范围内,表明两者无明显差异(见表8),所建立的一测多评法可以用于马鞭草质量评价研究。

表8 各成分含量测定结果Table 8 Results of content determination of various constituents
2.5 标准汤剂量值传递研究
2.5.1 马鞭草标准汤剂制备
按《中药配方颗粒质量控制与标准制定技术要求》制备18批马鞭草标准汤剂,取马鞭草饮片各100 g,煎煮2次,一煎加水12倍,浸泡30 min,武火煮沸,文火保持微沸30 min,用350目筛网趁热过滤;二煎加水10倍,武火煮沸,文火保持微沸25 min,用350目筛网趁热过滤;合并2次煎液,65 ℃真空减压浓缩至100 mL,冷冻干燥即得。
2.5.2 马鞭草标准汤剂含量、出膏率测定及转移率计算
马鞭草标准汤剂批号对应饮片批号分别为B1~B18,计算出膏率(出膏率=冻干粉重量/饮片重量×100%),18批马鞭草标准汤剂出膏率波动范围为10.94%~22.25%,平均出膏率为16.68%;测定毛蕊花糖苷、5-羟基马鞭草苷、马鞭草苷含量,并计算转移率(外标法)[13](见表9)。毛蕊花糖苷转移率的波动范围为7.9%~36.3%;5-羟基马鞭草苷转移率的波动范围为35.0%~113.4%;马鞭草苷转移率的波动范围为52.1%~109.2%。

表9 马鞭草标准汤剂出膏率、含量及转移率Table 9 Dry extract rate,content and transfer rate of Verbenae Herba standard decoction
3 讨论与结论
马鞭草样品特征图谱等度洗脱无法实现基线分离,故采用梯度洗脱,在前期预实验中,本研究对比了甲醇-水、乙腈-水、乙腈-0.1%磷酸溶液、乙腈-0.05%磷酸溶液等流动相,最终选定乙腈-0.05%磷酸溶液为流动相,在上述色谱条件下,色谱峰分离度、理论板数及峰型等参数均较好。本研究借助单因素实验系统考察了提取溶剂、提取方法及时间、料液比对提取效率的影响,结果显示,当提取溶剂为80%甲醇、料液比为1∶50(g/mL)、回流提取120分钟时,提取效率最高,故以上述方法为马鞭草饮片的提取方法。
中药多指标成分整体质量控制已成为中药质量控制必然发展趋势[14],特征/指纹图谱是一种有效的质量评价方法,将其与一测多评分析方法相结合可实现质量评价的整体性和准确性。本研究建立了马鞭草饮片UPLC特征图谱,确定了5个特征共有峰并指认了其中3个成分,分析时间比HPLC有明显缩短,且色谱条件可用于5-羟基马鞭草苷、马鞭草苷及毛蕊花糖苷含量测定。对特征峰峰面积进行主成分分析,该主成分综合评价模型进一步完善了马鞭草质量评价分析方法。
2020年《中国药典》中马鞭草的含量测定指标为齐墩果酸和熊果酸总量,均为三萜类化合物,极性小,水溶性较低,前期实验研究结果显示,马鞭草标准汤剂中齐墩果酸和熊果酸的总含量低于2.0 mg/g,转移率低于6%。因此,基于特征图谱,建立了5-羟基马鞭草苷、马鞭草苷及毛蕊花糖苷3种成分含量一测多评测定方法。马鞭草的极性成分以环烯醚萜类物质为主,其中马鞭草苷和5-羟基马鞭草苷为含量最高的主要成分,标准汤剂转移率较高。部分批次转移率超100%,而5-羟基马鞭草苷、马鞭草苷总量的总转移范围在56.6%~91.2%。可能原因为同类成分在加热煎煮等过程中发生结构转化。18批马鞭草标准汤剂中毛蕊花糖苷、5-羟基马鞭草苷和马鞭草苷含量和转移率批间存在差异,可能与饮片含量、饮片的质地有关,部分饮片颜色较浅,存放时间较久,炮制时,碎屑较多,可能是导致其含量和转移率波动较大的原因。所选的3种成分在马鞭草中含量较高,在抗氧化、保肝、保护神经、保护心脏及脑缺血、抗炎等方面具有较好的药理活性,是马鞭草发挥药效的主要物质基础之一。
一测多评法测定结果与外标法测定结果无明显差异,可用于马鞭草中多种成分定量分析。UPLC特征图谱结合多指标成分一测多评定量分析法可为马鞭草及马鞭草中药制剂工艺研究和质量评价提供参考。
