玉米禾谷镰孢菌穗腐病抗性基因组选择研究
2023-08-27周广飞高夕全
周广飞 高夕全
摘要:禾谷镰孢菌穗腐病是玉米种植区普遍发生的重要病害之一,探究基因组选择在禾谷镰孢菌穗腐病抗性遗传改良中的应用潜力,有助于选育抗病品种。以玉米自交系DH4866和T877组配的重组自交系群体,对玉米禾谷镰孢菌穗腐病抗性进行基因组选择研究,分析数据模型、群体大小、标记密度、显著位点和遗传效应对预测准确性的影响。结果表明,在5种数据模型中,gBLUP模型对玉米禾谷镰孢菌穗腐病抗性具有较高的预测能力。预测准确性随着训练群体样本大小的增加而提高,当训练群体占群体总数80%时,预测准确性达到最高。当标记数量达到500时,即可获得相对较高的预测准确性。相对于随机效应模型,将显著位点作为固定效应没有显著提高预测准确性。相对于仅考虑加性效应,当在模型中考虑加加上位性效应时,预测准确性并没有得到显著提高。研究结果可为玉米禾谷镰孢菌穗腐病抗性的遗传改良提供理论参考。
关键词:玉米;禾谷镰孢菌穗腐病;基因组选择;预测准确性;遗传改良
中图分类号:S435.131.4+9 文献标志码:A
文章编号:1002-1302(2023)14-0065-05
玉米穗腐病是由多种病原菌引起的,世界玉米种植区普遍发生的一种真菌性病害,其中由禾谷镰孢菌引起的穗腐病是我国玉米穗腐病的主要类型之一[1-2]。禾谷镰孢菌穗腐病不仅可以造成玉米籽粒产量和商业品质下降,导致经济损失,而且会产生脱氧雪腐镰孢菌烯醇和玉米赤霉烯酮等毒素,严重威胁人畜健康[3-4]。由于镰孢菌种类多样,很容易适应多种环境和不同杀菌剂,导致通过栽培管理和化学手段防控效果不理想,使用抗病品种是降低玉米禾谷镰孢菌穗腐病发生的有效手段[5-6]。因此,选育抗病品种是减少玉米禾谷镰孢菌穗腐病导致的产量损失、提高籽粒商業品质的重要手段。但玉米禾谷镰孢菌穗腐病抗性属于复杂数量性状,易受环境影响,利用传统的育种方法进行遗传改良效率较低[1]。
基因组选择是随着现代测序技术发展而来的,一种利用覆盖全基因组高密度分子标记进行预测的育种方法,该方法主要是通过已知表型和基因型的训练群体,构建统计模型,预测仅已知基因型的育种群体的育种值,可以在减少田间工作量的同时,提高选择效率[7]。基因组选择是利用全基因组的标记进行选择,可以估算出所有的遗传效应,解释全部的遗传变异,即便是标记的效应微效,也能估算出来,比较适合用于由微效多基因控制的复杂数量性状的遗传改良[8]。作为一种新的育种策略,与传统表型选择和分子标记辅助选择相比,基因组选择具有选择准确度高、世代间隔短、育种效率高等优点,目前已经被越来越多地应用于农作物的育种领域[9]。
目前已有关于基因组选择在玉米禾谷镰孢菌穗腐病抗性方面的应用,均是以国外的玉米种质为研究对象。Han等以130份硬粒型和114份马齿型欧洲玉米自交系为试验材料,利用全基因组的 23 797 个标记对玉米禾谷镰孢菌穗腐病抗性进行基因组选择,预测准确性可达0.50[10]。Galiano-Carneiro等分别利用巴西与欧洲玉米自交系构建的多亲本群体和2个欧洲地方品种构建的双单倍体群体,发现将与玉米禾谷镰孢菌穗腐病抗性显著关联的主效位点作为固定效应加入基因组选择数据模型,可以提高预测准确性,但预测准确性受训练群体与测试群体的亲缘关系影响[11-12]。
利用基因组选择技术有助于玉米禾谷镰孢菌穗腐病抗性的遗传改良,但基因组选择预测准确性受多种因素影响,如数据模型、群体大小和标记密度等。利用不同的遗传群体,对这些影响因素进行深入研究,有利于提高预测准确性,加快育种进程。本研究将利用由我国骨干玉米自交系DH4866×T877组配的重组自交系群体,通过3个环境的抗病性鉴定,分析数据模型、群体大小、标记密度、显著位点和遗传效应对基因组选择预测准确性的影响,以期为玉米禾谷镰孢菌穗腐病抗性的遗传改良提供理论参考。
1 材料与方法
1.1 试验材料与田间试验
试验材料为由DH4866×T877组配的重组自交系群体,包含204个家系[13],其中DH4866是登海1号的母本,以7922×Ye478为基础材料选育,T877是苏玉30的父本,以美国杂交种78599×E28为基础材料选育[14]。
重组自交系群体田间抗性鉴定于2019年在海南三亚(108°E,18°N)、江苏南通(120°E,31°N)和河南新乡(113°E,35°N)等3个环境进行。每个环境的田间试验均采用随机区组设计,重复2次,单行区,行长为3.0 m,行距为0.6 m,种植密度为 6.5万株/hm2。
1.2 田间接种与表型鉴定
玉米吐丝10 d后,首先用锥子在第1个玉米果穗上中下部各制造1个伤口,扎破1~2粒玉米,不刺到穗轴,然后用连续注射器在每个伤口各注射200 μL禾谷镰孢菌菌液(1×106个孢子/mL),喷水保湿[15]。待玉米成熟后,调查果穗的发病情况。
抗病性鉴定根据发病面积占果穗面积的比例进行分级,参考Reid等的方法分为1~7级:1级,没有发病;2级,发病面积占果穗面积的1%~3%;3级,发病面积占果穗面积的4%~10%;4级,发病面积占果穗面积的11%~25%;5级,发病面积占果穗面积的26%~50%;6级,发病面积占果穗面积的51%~75%;7级,发病面积占果穗面积的76%~100%[16]。
1.3 基因型分析与QTL定位
利用Axiom Maize56K SNP Array分析重组自交系群体的基因型,获得了9 780个高质量的单核苷酸多态性(SNP),将共分离的SNP标记视为1个bin,利用JoinMap 4.0软件,将bin作为标记,利用Kosambi计算标记间的遗传距离,构建的遗传图谱包含1 868个bin,遗传图谱总长3 081.84 cM,平均遗传距离为1.65 cM[13]。
利用Windows QTL Cartographer 2.5软件中的复合区间作图法进行数量性状基因座(QTL)分析,在三亚、南通、新乡以及这3个环境的最佳线性无偏预测(BLUP)值中,共鉴定到11个控制玉米禾谷镰孢菌穗腐病抗性的QTL[15]。
1.4 全基因组选择分析
为研究不同因素对玉米禾谷镰孢菌穗腐病抗性基因组选择的影响,采用多种策略分析基因组选择的预测准确性。
(1)为研究不同数据模型对预测准确性的影响,选择rrBLUP、gBLUP、BayesA、BayesB和BayesC等5种数据模型进行基因组选择。利用R语言中rrBLUP包构建rrBLUP模型[17],GAPIT包构建gBLUP模型[18],BGLR包构建BayesA、BayesB和BayesC模型[19]。
(2)为研究群体大小对预测准确性的影响,设置不同的群体大小作为训练群体进行基因组选择,选取群体中10%、20%、30%、40%、50%、60%、70%、80%、90%的材料作为训练群体,其余的材料作为测试群体,重复100次。
(3)为研究标记密度对预测准确性的影响,设置不同的标记密度进行基因组选择,标记密度设置为25、50、100、200、300、400、500、600、700、800、900、1 000、1 200、1 400、1 600、1 868(ALL)个标记,每个标记密度中的标记都是随机选取,重复100次。
(4)为研究显著标记对预测准确性的影响,将11个QTL作为固定效应加入数据模型。
(5)为研究遗传效应对预测准确性的影响,将基因加加上位性效应加入数据模型。
采用5倍交叉验证估计预测准确性,即群體中80%的材料作为训练群体,其余20%的材料作为测试群体,重复100次。预测的准确性定义为预测值与真实值之间的皮尔逊相关系数[20]。
2 结果与分析
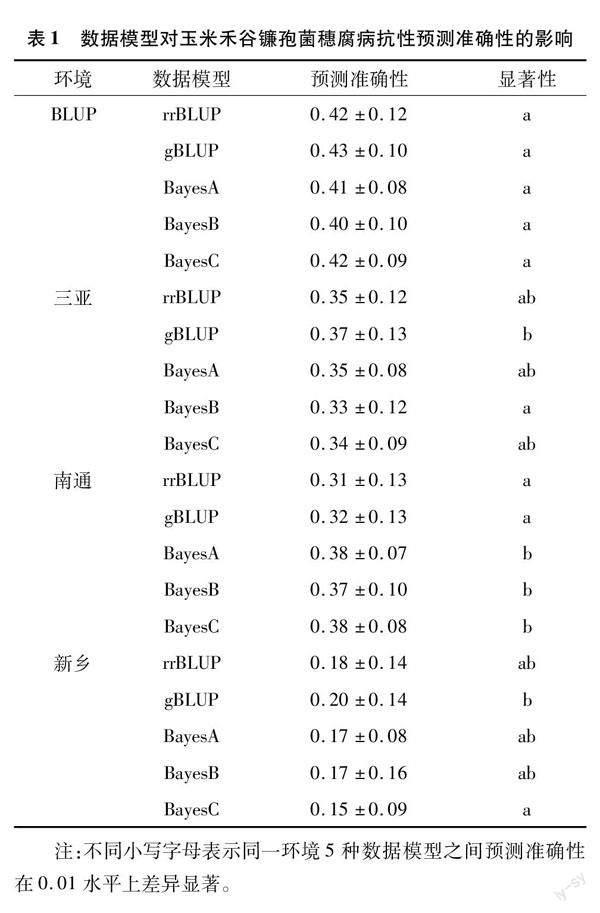
2.1 数据模型对预测准确性的影响
由表1可知,在同一环境中,5种数据模型对玉米禾谷镰孢菌穗腐病抗性的预测准确性存在差异。利用3个环境的BLUP值作为表型,5种数据模型的预测准确性之间没有显著差异,在三亚和新乡环境中,gBLUP的预测准确性最高,在南通试验点,BayesA、BayesB和BayesC的预测准确性显著优于rrBLUP和gBLUP。总体看来,在本研究中gBLUP的预测准确性最优。利用相同的数据模型,不同环境间的预测准确性存在明显差异,利用BLUP值预测的准确性最高,而利用新乡的表型数据,预测准确性最差,说明玉米禾谷镰孢菌穗腐病抗性易受环境影响,通过多环境的抗病性鉴定,可以提高基因组选择的预测准确性。因此,在后续分析中,利用gBLUP模型和群体的BLUP值作为表型进行基因组选择。
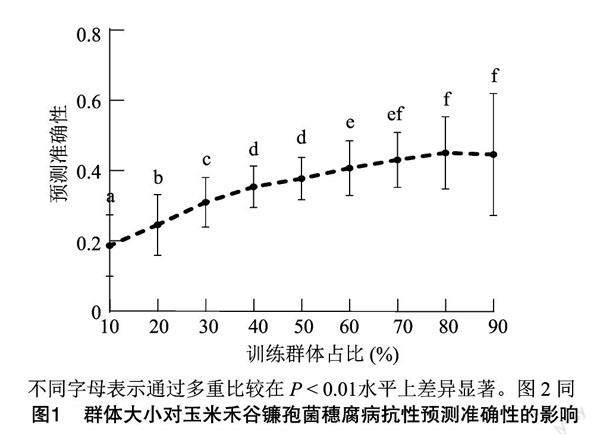
2.2 群体大小对预测准确性的影响
由图1可知,随着训练群体逐渐增大,基因组选择预测准确性呈先上升后稳定的趋势。当训练群体由10%上升到60%时,预测的准确性呈快速升高的趋势,由0.19上升到0.41,当训练群体为80%时,预测准确性达到最大值0.45,到90%时,预测准确性略微下降,虽然差异不显著,但标准差明显增加,稳定性降低。
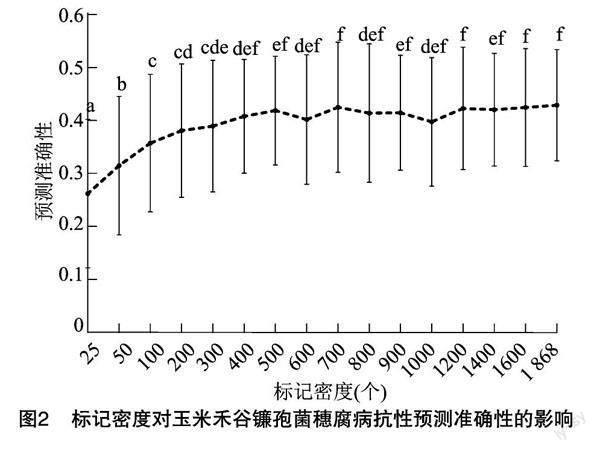
2.3 标记密度对预测准确性的影响
由图2可知,随着标记密度的增加,基因组选择预测准确性呈先上升后稳定的趋势。随机选取的标记密度从25个增加到500个,预测的准确性快速提高,由0.26上升到0.42,之后随着标记密度的增加,预测准确性有所提高,但差异不显著。
2.4 显著位点对预测准确性的影响
由图3-A可知,将11个抗性QTL峰值的标记作为固定效应进行基因组选择,相对于随机效应,预测准确性有所提高,差异不显著,但极显著高于随机选择11个标记作为固定效应的预测准确性。
2.5 遗传效应对预测准确性的影响
由图3-B可知,与仅考虑加性效应的统计模型相比,当统计模型考虑加加上位性效应后,预测准确性略微下降,但差异不显著,主要原因可能是在重组自交系群体中,玉米禾谷镰孢菌穗腐病抗性的广义遗传率较高(69.84%)[15],基因加性效应是表型变异的主要因素。
3 讨论
数据模型是基因组选择的核心,合适的数据模型将会显著提高预测准确性。由于不同数据模型的先验假设和对标记效应分布的假设不同,导致预测准确性不同。根据假设测验不同,主要分为BLUP和Bayes[9,21-22]。BLUP类假定所有分子标记具有相同的遗传方差,主要包括gBLUP和rrBLUP。gBLUP是利用分子标记信息构建基因组关系矩阵[WTHX]G,估算个体育种值,而rrBLUP是将标记效应作为随机效应且服从正态分布,利用线性混合模型估算每个标记的效应值,然后将每个标记效应相加,估算个体育种值。实际上,全基因组中仅有少数分子标记有效应,大多数标记是无效应的,而Bayes法是将标记效应的方差假定为某种先验分布,例如:BayesA假设每个分子标记都有效应且服从正态分布,遗传方差服从尺度逆卡方分布;BayesB假设少数分子标记有效应,遗传方差服从逆卡方分布;BayesC将BayesB中的参数π作为未知参数,假定其服从U(0,1)的均匀分布,并假设具有效应的分子标记,其遗传差不同。与BLUP类模型相比,Bayes类模型具有更多的参数估计,计算量大,耗时长。本研究利用rrBLUP、gBLUP、BayesA、BayesB和BayesC进行玉米禾谷镰孢菌穗腐病抗性基因组选择,利用3个环境的BLUP值作为表型,5种数据模型中,gBLUP模型的预测准确性最高,但与其余4种模型之间没有显著差异。不同环境之间,利用相同的数据模型和分子标记,预测准确性之间存在显著差异,说明玉米禾谷镰孢菌穗腐病抗性易受环境影响。这与玉米拟轮枝镰孢菌穗腐病抗性基因组选择的结果[23]类似。
在研究群体大小对预测准确性影响时,通常会考虑训练群体与测试群体规模的比例。本研究发现当训练群体的个体数量是测试群体的4倍时,预测的准确性达到最大值。该结果与前人的研究结果有所不同,Cao等对玉米复合焦油斑病抗性研究时,当训练群体个体数量与测试群体一样时,预测准确性达到最大值[24],而Liu等对玉米株型、产量等相关性状进行基因组选择时,当训练群体大小是测试群体3倍时,实现最高水平的预测。这种情况可能是使用群体和目标性状不同导致的[25]。
标记密度是影响预测准确性的另一个重要因素。利用不同的群体,当预测准确性达到最高时,所对应的标记密度各不相同,可能由不同的群体遗传多样性造成的。一般情况下,自然群体遗传结构复杂,标记间连锁不平衡程度较高,因此需要大量的分子标记才能保证至少有1个标记与性状相关位点处于连锁不平衡状态,以便实现高水平的预测准确性[7]。然而对于双亲群体,由于群体结构清晰,以及在群体构建过程中只产生了有限的重组,大量亲本染色体片段被传递给子代,因此中等密度的分子标记就可以保证至少有1个标记与性状相关位点处于连锁不平衡状态,并且极有可能获得较高水平的预测[26]。在玉米拟轮枝镰孢菌穗腐病抗性基因组选择分析中,Guo等以包含509份玉米自交系组成的自然群体为材料,发现需要4 000个标记,可以获得一个较高水平的预测(0.36)。本研究利用的重组自交系群体,利用500个标记,即可获得较好的预测(0.42),之后通过增加标记数量对预测准确性的提高并没有明显的帮助[23]。
在基因组选择随机模型中,假设所有标记效应的分布具有相同的方差,这样会导致在估计亲缘关系矩阵时,与性状显著关联标记的作用被稀释,然而将这些与性状显著关联的标记作为固定效应加入模型中,其效应分布的方差与作为随机效应的标记不同,可以解释部分遗传方差,进而提高预测的准确性[27]。利用全基因组关联分析检测与玉米禾谷镰孢菌穗腐病显著关联的抗性位点,将效应值较高的位点(表型变异贡献率>5%)作为固定效应,可以显著提高预测准确性[11-12]。前期利用连锁分析鉴定到的11个QTL[15],本研究将这11个QTL的峰值标记作为固定效应,与随机效应相比,预测准确性尽管有所提高,但差异不显著,可能是由于QTL区间较大,峰值的标记并不能代表这个QTL的功能位点,也可能由于标记效应较小,不能反映出足够多的遗传方差[27-28]。
参考文献:
[1]Gaikpa D S,Miedaner T. Genomics-assisted breeding for ear rot resistances and reduced mycotoxin contamination in maize:methods,advances and prospects[J]. Theoretical and Applied Genetics,2019,132(10):2721-2739.
[2]段燦星,王晓鸣,宋凤景,等. 玉米抗穗腐病研究进展[J]. 中国农业科学,2015,48(11):2152-2164.
[3]Zhou M,Yang L J,Shao M H,et al. Effects of zearalenone exposure on the TGF-β1/Smad3 signaling pathway and the expression of proliferation or apoptosis related genes of post-weaning gilts[J]. Toxins,2018,10(2):49.
[4]Pinton P,Oswald I P. Effect of deoxynivalenol and other type B trichothecenes on the intestine:a review[J]. Toxins,2014,6(5):1615-1643.
[5]Talas F,Mcdonald B A. Significant variation in sensitivity to a DMI fungicide in field populations of Fusarium graminearum[J]. Plant Pathology,2015,64(3):664-670.
[6]Munkvold G P. Cultural and genetic approaches to managing mycotoxins in maize[J]. Annual Review of Phytopathology,2003,41(1):99-116.
[7]Meuwissen T H E,Hayes B J,Goddard M E. Prediction of total genetic value using genome-wide dense marker maps[J]. Genetics,2001,157(4):1819-1829.
[8]Bernardo R,Yu J M. Prospects for genomewide selection for quantitative traits in maize[J]. Crop Science,2007,47(3):1082-1090.
[9]Voss-Fels K P,Cooper M,Hayes B J. Accelerating crop genetic gains with genomic selection[J]. Theoretical and Applied Genetics,2019,132(3):669-686.
[10]Han S,Miedaner T,Utz H F,et al. Genomic prediction and GWAS of Gibberella ear rot resistance traits in dent and flint lines of a public maize breeding program[J]. Euphytica,2018,214(1):6.
[11]Galiano-Carneiro A L,Kessel B,Presterl T,et al. Multi-parent QTL mapping reveals stable QTL conferring resistance to Gibberella ear rot in maize[J]. Euphytica,2020,217(1):1-13.
[12]Gaikpa D S,Kessel B,Presterl T,et al. Exploiting genetic diversity in two European maize landraces for improving Gibberella ear rot resistance using genomic tools[J]. Theoretical and Applied Genetics,2021,134(3):793-805.[HJ1.9mm]
[13]Li P C,Zhang Y Y,Yin S Y,et al. QTL-by-environment interaction in the response of maize root and shoot traits to different water regimes[J]. Frontiers in Plant Science,2018,9:229.
[14]Zhou G F,Hao D R,Xue L,et al. Genome-wide association study of kernel moisture content at harvest stage in maize[J]. Breeding Science,2018,68(5):622-628.
[15]Zhou G F,Li S F,Ma L,et al. Mapping and validation of a stable quantitative trait locus conferring maize resistance to Gibberella ear rot[J]. Plant Disease,2021,105(7):1984-1991.
[16]Reid L M,Mather D E,Bolton A T,et al. Evidence for a gene for silk resistance to Fusarium graminearum Schw. ear rot of maize[J]. Journal of Heredity,1994,85(2):118-121.
[17]Endelman J B. Ridge regression and other kernels for genomic selection with R package rrBLUP[J]. The Plant Genome,2011,4(3):250-255.
[18]Lipka A E,Tian F,Wang Q S,et al. GAPIT:genome association and prediction integrated tool[J]. Bioinformatics,2012,28(18):2397-2399.
[19]Pérez P,de los Campos G. Genome-wide regression and prediction with the BGLR statistical package[J]. Genetics,2014,198(2):483-495.
[20]Crossa J,Pérez P,Hickey J,et al. Genomic prediction in CIMMYT maize and wheat breeding programs[J]. Heredity,2014,112(1):48-60.
[21]Wang X,Xu Y,Hu Z L,et al. Genomic selection methods for crop improvement:current status and prospects[J]. The Crop Journal,2018,6(4):330-340.
[22]Heslot N,Yang H P,Sorrells M E,et al. Genomic selection in plant breeding:a comparison of models[J]. Crop Science,2012,52(1):146-160.
[23]Guo Z F,Zou C,Liu X G,et al. Complex genetic system involved in Fusarium ear rot resistance in maize as revealed by GWAS,bulked sample analysis,and genomic prediction[J]. Plant Disease,2020,104(6):1725-1735.
[24]Cao S L,Loladze A,Yuan Y B,et al. Genome-wide analysis of tar spot complex resistance in maize using genotyping-by-sequencing SNPs and whole-genome prediction[J]. The Plant Genome,2017,10(2):plantgenome2016.10.0099.
[25]Liu X G,Wang H W,Wang H,et al. Factors affecting genomic selection revealed by empirical evidence in maize[J]. The Crop Journal,2018,6(4):341-352.
[26]Lorenzana R E,Bernardo R. Accuracy of genotypic value predictions for marker-based selection in biparental plant populations[J]. Theoretical and Applied Genetics,2009,120(1):151-161.
[27]Moore J K,Manmathan H K,Anderson V A,et al. Improving genomic prediction for pre-harvest sprouting tolerance in wheat by weighting large-effect quantitative trait loci[J]. Crop Science,2017,57(3):1315-1324.
[28]Spindel J E,Begum H,Akdemir D,et al. Genome-wide prediction models that incorporate de novo GWAS are a powerful new tool for tropical rice improvement[J]. Heredity,2016,116(4):395-408.
收稿日期:2022-10-12
基金項目:江苏省种业振兴揭榜挂帅项目(编号:JBGS[2021]054、JBGS[2021]009);江苏省南通市科技项目(编号:JC2021153、MS22021039);江苏沿江地区农业科学研究所学科建设专项基金(编号:YJXK[2021]201)。
作者简介:周广飞(1988—),男,山东济宁人,博士,助理研究员,从事玉米遗传育种研究。E-mail:gfzhou88@jaas.ac.cn。
