芯片和重测序在猪遗传结构研究中的应用比较
2023-07-31赵雪艳朱晓东耿立英张传生王继英
杨 晴,巩 静,赵雪艳,朱晓东,耿立英,张传生*,王继英*
(1.河北科技师范学院动物科技学院,秦皇岛 066600;2.山东省农业科学院畜牧兽医研究所 山东省畜禽疫病防治与繁育重点实验室,济南 250100;3.农业农村部畜禽生物组学重点实验室,济南 250100;4.枣庄黑盖猪养殖有限公司,枣庄 277100)
随着高密度芯片和测序技术的高速发展,生物全基因组范围内检测出的标记数量逐渐增多,大量的遗传标记信息使基因组遗传变异分析得以更为准确和精准的实施。当前,SNP芯片与测序技术已成为动植物进行遗传变异信息分析工作的主要工具,被广泛应用于遗传多样性分析[1-2]、选择信号检测[3-4]、全基因组关联分析[5-6]、基因组选择等[7-8]。SNP芯片具有自动化、成本低、效率高等优点,但其存在检测位点较少、特异性强、无法发现新功能位点等缺点[9]。测序不受参考基因组的限制,甚至可通过提高测序深度来获得所测样本的全部遗传变异信息,包括覆盖低、中、高密度甚至全基因组范围内的所有已知或未知的SNP位点信息[10],但其存在数据量大、分析复杂、成本较高的缺点[11]。实际研究中,还是要根据研究目的选择适当的分型方法。虽然增加SNP检测密度会提高分析结果的准确度,但在实际应用中,高密度分型会带来高昂的经济成本,极大地限制了高密度标记在基因组遗传变异分析中的应用空间,所以根据研究目的探索适当的标记密度和经济的分型方法,保证分析结果的准确性,成为近年来SNP标记分析的热点研究内容之一。
已有报道显示,低密度面板的基因组选择,通过基因型填充等方法能够达到中高密度,甚至测序数据相似的基因组预测精确程度,是一种低成本且高效的遗传评估方法[12-14]。但是分析不同分型方法或不同SNP密度对全基因组遗传变异分析结果是否存在影响且影响是否较大的报道仍较少。因此,本研究以35头枣庄黑盖猪的高密度SNP芯片数据和重测序SNP数据为基础,利用重测序信息构建不同密度的SNP面板,以探究不同SNP分型方法和不同SNP密度对遗传变异分析的影响,找到适用于遗传变异分析的低成本、高效的分型方法和SNP密度,为今后猪及其他畜禽遗传特性分析中适宜的基因分型技术和标记密度的选择提供重要参考。
1 材料与方法
1.1 样本来源
本研究所用的35头枣庄黑盖猪均采自山东省枣庄黑盖猪养殖有限公司,包括16头母猪和19头公猪。采集试验猪耳组织样品存放于装有75%酒精的2 mL冻存管内,放入-20 ℃低温冰箱中保存备用。
1.2 DNA提取与质检
取样本耳组织0.5 g左右,采用血液/细胞/组织基因组DNA提取试剂盒(DP304,TIANGEN公司,北京)进行基因组DNA的提取。利用NanoDrop 2000和琼脂糖凝胶电泳对DNA的浓度和质量进行检测,浓度>50 ng·μL-1,1.8 使用CAUPorcineSNP50芯片(北京康普森生物技术有限公司)对35个个体进行SNP分型,SNP检出率平均为97.97%。基于华大-MGISEQ-T7技术测序平台,利用双末端测序(paired-end)的方法对35个个体进行基因组重测序,平均测序深度为13X,Q20为98.18%。原始数据质控后,使用BWA软件[15]的BWA-MEN算法将质控数据与参考基因组(Ensembl Sus Scrofa11.1)进行比对,使用GATA[16]进行重比对,最后使用Samtools软件[17]和Bcftools软件[18]检测基因组范围内的SNP。 使用Plink(V1.90)[19]对SNP芯片和重测序中的数据按如下标准进行质量控制,标准如下:1)仅保留位于常染色体上的SNP位点;2)芯片数据删除检出率(call rate)<90%的SNP位点,重测序数据删除检出率<95%的SNP位点;3)删除检出率<90%的个体;4)删除最小等位基因频率(MAF)<0.05的SNP位点。 基于重测序检测的SNP位点,利用R语言CVrepGPAcalc包(https://github.com/SmaragdaT/CVrep/)构建不同密度的SNP面板[20],依据SNP芯片密度共设计了3个梯度,分别为34K、340K和3 400K。面板的构建有两种方法,第一种是在整个基因组中随机抽样来选择SNP,第二种是根据特定步长的物理距离均匀的选择SNP。其中,34K面板选择两种方法分别进行构建,340K和3 400K面板均采用第二种方法进行构建。 使用Plink(V1.90)计算群体的最小等位基因频率(minor allele frequency, MAF)、观察杂合度(observed heterozygosity,HO)、期望杂合度(expected heterozygosity,HE)、群体内遗传距离等遗传多样性指标,使用Plink(V1.90)将数据格式转化为vcf格式,再利用vcf2phylip和Phylip通过邻接法(neighbor-joining, NJ)构建系统发生树[21-22],最后利用FigTreev1.4.4软件(http://tree.bio.ed.ac.uk/software/figtree/)将计算结果可视化。使用Plink(V1.90)计算状态同源距离(identity by descent distance, IBS距离),随后计算个体间遗传距离(1-IBS距离),并利用BioLadder在线软件(https://www.bioladder.cn/web/#/chart/6)绘制个体间遗传距离热图。 使用R语言CMplot软件包对SNP在染色体上的分布进行可视化,使用R语言detectRUNS软件包[23]对基因组进行长纯合片段(runs of homogeneity,ROH)检测并计算各分组内的群体内近交系数(FROH),参数设置[24-26]为:SNP密度最小为每1 000 kb必须有1个SNP;连续两个SNPs的间隔最大为1 000 kb;滑窗大小为50个SNPs;ROH滑窗中允许有1个SNP位点为杂合;ROH滑窗中允许有5个SNPs位点缺失;滑动窗口重叠比例至少为5%;ROH最少个数为40个SNPs。 利用CAUPorcineSNP50 芯片和基因组重测序对35头枣庄黑盖猪进行基因组SNP检测,分别获得了43 832个和31 437 418个SNPs位点。芯片的SNP检出率平均为0.979 8,重测序的检出率平均为0.997 0。各质控条件下芯片和重测序数据SNP位点的详细剔除数量见表1。经过数据质控后,芯片和测序数据剩余位点的比例分别为78.69%和65.76%。 表1 SNP质控结果汇总Table 1 Summary of SNP quality control results 通过质控标准的芯片SNP位点个数为34 494个。依据芯片密度(34K)设置梯度,以重测序数据为“原材料”构建不同密度SNP面板。芯片和各密度SNP面板的SNP位点数目、MAF和相邻SNP间距详见表2。可以看出,芯片标记MAF均值为0.292,高于测序各组标记的MAF均值(0.244~0.245)。密度同为34K的3组相比,芯片SNP间距均值最大(70 809.82 bp),均匀34K的次之(65 819.90 bp),随机34K的最小(63 359.20 bp)。但是,随机34K组SNP间距的标准差最大(80 185.61 bp),远高于芯片(57 626.21 bp)和均匀34K(1 771.16 bp)。综合来看,芯片的SNP位点在染色体上的分布均匀度介于随机34K和均匀34K之间。与图1密度分布图所示结果一致。不同密度测序SNP面板(均匀34K、均匀340K和均匀3 400K)相比较,均匀34K的SNP间距均值约为均匀340K的10倍,基本与构建面板时采用的步长大小(10×)相一致,标准差大小随SNP密度的增加而减小。 表2 芯片和各测序面板SNP数目、最小等位基因频率和间距Table 2 SNP number, MAF and space of adjacent SNPs of array and sequencing panels 利用芯片和各测序SNP面板的SNP标记分析枣庄黑盖猪的遗传多样性结果见表3。可以看出,利用芯片SNP标记分析的HO、HE、遗传距离均高于测序各组,利用各测序面板SNP标记分析的HO、HE、遗传距离基本相同,特别是均匀分布的3组SNP(34K、340K和3400K)的遗传多样性指标更为接近。图2展示了使用芯片和测序各组数据分析的35头枣庄黑盖猪样本间遗传距离矩阵热图,与表3结果一致,芯片与随机34K及均匀分布SNP组间的差别最为明显。 A. 芯片;B. 随机34K;C. 均匀34K。矩阵中每一个小方格代表样本两两之间的遗传距离值,该值越大越接近紫色,越小越接近黄绿色A. Array; B. Random 34K; C. Even 34K. Each small square in the matrix represents the genetic distance value between two samples, the larger the value, the color is closer to purple, and the smaller the value, the color is closer to yellow-green图2 样本间遗传距离热图Fig.2 Heat map of genetic distance between samples 表3 芯片和各测序面板遗传多样性参数值Table 3 Values of genetic diversity analyzed based on array and sequencing panels 利用芯片和各测序SNP面板的SNP标记构建了枣庄黑盖猪群体邻接法系统发生树,详见图3。系统发生树是表示个体间亲缘关系的树状图,相同分支上的个体具有相近亲缘关系,为同一个家系。可以看出,基于芯片和各测序SNP面板的SNP标记构建的系统发生树均将35头枣庄黑盖猪划分为3大分支,每个大分支又可进一步细分成1~3个小分支。仔细对比分支上的个体,芯片与随机34K、芯片与3组均匀SNP数据均存在一定的差别,而3个均匀分布的SNP数据(34K、340K和3 400K)构建的系统发生树基本一致。 A. 芯片;B. 随机34K;C. 均匀34K;D. 均匀340KA. Array; B. Random 34K; C. Even 34K; D. Even 340K图3 邻接法构建的系统发生树Fig.3 Phylogenetic trees constructed by neighbor-joining method 利用芯片和各测序SNP面板的SNP标记分析了枣庄黑盖猪ROH和基因组近交系数,详见表4。可以看出,芯片与随机34K相比,芯片检测的ROH数目少(723vs. 784),但ROH长度大(14.86 Mbvs. 12.85 Mb),二者的FROH相近(0.125vs.0.127);均匀34K与随机34K相比,均匀34K数据检测到ROH数目更多(789vs. 784),长度更大(13.51 Mbvs. 12.85 Mb),FROH近交系数更高(0.134vs. 0.127)。3个均匀分布的数据组相比,随着标记密度增加,检测的ROH数目逐渐增多,ROH长度逐渐降低,估计的FROH近交系数也逐渐增加。 表4 芯片和各测序面板ROH及基因组近交系数值Table 4 ROH and genomic inbreeding coefficients based on array and sequencing panels 单核苷酸多态性(SNPs)是人类和其他动物可遗传的变异中最常见的一种,在基因组中广泛存在,作为第三代分子标记在畜禽遗传多样性分析、选择信号检测、全基因组关联分析、基因组选择等方面发挥着重要作用。近来的研究表明,人类基因组上SNP总数可达3 800万个[27],目前已鉴定出的猪SNP已经超过四千余万个[28]。基因组测序可以获得所测样本的全部SNP信息,因此,WGS数据有望可以用来更好地估计个体之间的真实关系[29]。SNP芯片仅包含了鉴定出的SNP位点的一个子集,SNP芯片的覆盖率和密度适当的情况下,在估计基因组关系、遗传多样性分析等方面与测序技术一样有价值[30]。 本研究中,35头枣庄黑盖猪基因组重测序共检测到3 143.7万个SNPs位点,经过数据质控后,测序数据剩余位点的比例(65.76%)小于芯片数据(78.69%),这与基因组重测序检测到的SNPs中含有大量的(6 976 769个,占位点总数的22.19%)低MAF位点(MAF<0.05)有关。与本研究结果一致,Wang 等[31]、Eynard等[29]在对大约克猪、荷斯坦牛的基因组测序数据分析中也发现基因组测序包含了20%左右的低MAF(MAF<0.05)位点。与基因组测序相比,芯片基因组SNP在设计过程中,优先选择测序样本中发现的高MAF的SNP位点[32]。本研究所用的CAUPorcineSNP50 芯片整合现有重要经济功能基因公开报道的候选位点,并加入部分地方猪种全基因组重测序鉴定的特有SNP综合优化研制而成,所以该芯片SNP位点平均MAF值(0.292)高于各测序面板(0.244~0.245)。 利用芯片SNP标记分析的HO、HE、遗传距离等遗传多样性各指标值均高于测序各组,利用芯片SNP标记构建的系统发生树与测序各组也存在较大不同,而测序各组SNP标记分析的遗传多样性各指标值基本相同,构建的系统发生树基本相似。本研究结果说明,分型方法对遗传多样性、遗传距离和系统发生树分析存在影响。以往的研究表明,芯片SNP位点由于倾向于选择高MAF位点、位点群体代表性不全面等原因(即确定偏倚(ascertainment biases))会影响遗传多样性、群体分化、连锁不平衡等分析的结果[33-35]。据此推测,本研究中芯片与测序对遗传距离分析结果的不同可能是由于芯片和测序标记MAF差异所致。在测序方法下,不同SNP密度对遗传多样性、遗传距离和系统发生树分析结果影响较小,说明3.4万个标记已经能充分满足系统发生树分析所需的标记数量,增加标记数目和增加数据运算量并不能进一步提高遗传多样性和系统发生树的分析精确性。 本研究以重测序数据为“原材料”构建了不同密度SNP面板,利用芯片和各测序SNP面板的SNPs标记分析枣庄黑盖猪的遗传多样性、系统发生树和基因组近交系数。结果表明,利用芯片SNP标记分析的HO、HE、遗传距离等遗传多样性指标值均高于各测序组,利用芯片SNP标记构建的系统发生树与各测序组也存在较大不同,此外,芯片数据检测出的ROH长度较测序组大,基于ROH计算的近交系数偏小。各测序组的不同SNP密度对遗传多样性和系统发生树分析结果影响较小,但对ROH及基于ROH计算的基因组近交系数影响很大。因此,在研究初期进行试验设计时,要根据研究目的选择适宜的基因分型技术和标记密度,以降低成本和提高结果的准确性。1.3 基因分型和质控
1.4 不同密度SNP面板的构建
1.5 数据分析
2 结 果
2.1 SNP分型与质控

2.2 不同密度SNP面板的构建

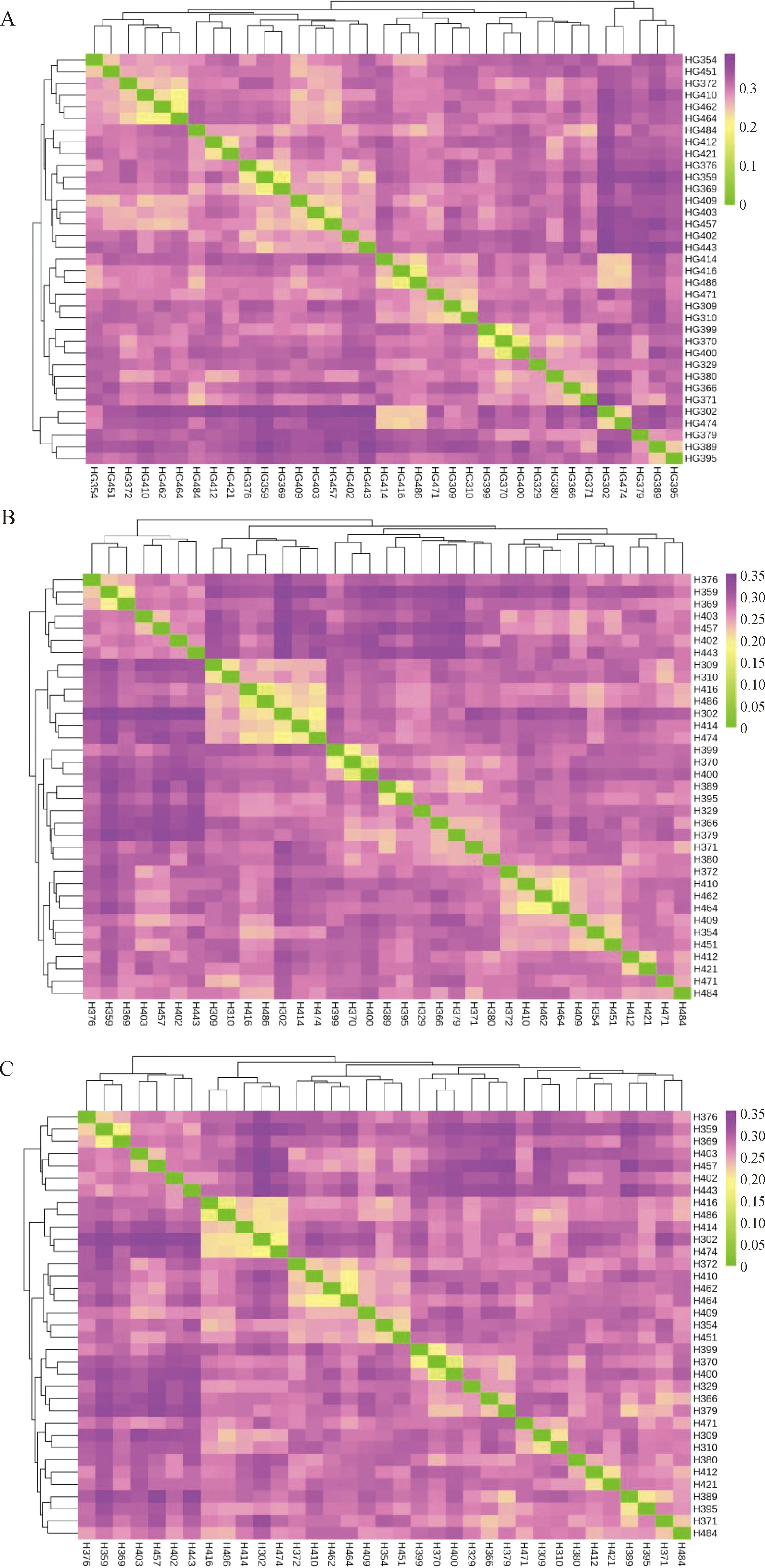
2.3 遗传多样性和遗传距离分析

2.4 系统发生树

2.5 基于ROH的基因组近交系数分析

3 讨 论
4 结 论
