基于全基因组重测序对晋汾白猪单核苷酸多态性位点鉴定和筛选
2023-07-31张万锋高鹏飞郭晓红蔡春波曹果清李步高
路 畅,董 磊,2,张万锋,高鹏飞,郭晓红,蔡春波,曹果清,李步高*
(1.山西农业大学动物科学学院,太谷 030801;2.山西省长治市潞城区现代农业发展中心,长治 047500)
中国地方猪种肉品质好、抗逆性强,但生长速度慢、饲养周期长使得其难以实现规模化和商业化饲养,因此,通过利用地方品种对国外猪种进行改良,快速开展新品种培育工作和评价,对猪种性能改进和提高、充分利用杂种优势和丰富现有种质资源具有重要意义。近年来,动物育种(尤其是品种改良)已经从群体水平进入了分子水平,主要通过利用测序技术在基因组水平获取更为准确的遗传信息,包括基因图谱构建、数量性状定位、标记辅助选择以及基因组选择等,为动物重要经济性状研究、起源进化以及疾病发生机制解析提供新的手段[1-2]。
随着测序技术的快速发展,以二代测序技术为主的高通量全基因组重测序技术推动了动植物单核苷酸多态性(single nucleotide polymorphism, SNP)的挖掘与注释工作[3-4]。全基因组重测序技术将基因组测序数据与参考基因组比对分析,能够获得包括单核苷酸多态性(SNP)等大量的遗传变异信息,为发掘畜禽种质特性、筛选影响畜禽生产性能的关键通路以及主效基因提供良好的平台[5]。SNPs作为基因组中最常见的遗传变异,被广泛应用于重要经济性状关键基因挖掘、种质鉴定、分子育种等方面[6-7]。在影响重要经济性状关键基因挖掘方面,Li等[3]对太湖猪、长白猪、杜洛克猪、滇南小耳猪和藏猪进行全基因组测序,共获得了132 078个太湖猪特异性的SNPs位点,并发现BMPR1B内含子上的雌激素应答元件ERE通过顺式调控作用影响猪的繁殖力;Wang等[8]将25头香猪基因组重测序数据与藏猪、梅山猪、杜洛克和约克夏的数据比较,筛选了3 985 444个特异性SNPs位点,鉴定了NR6A1和LTBP2基因的错义突变可能与香猪脊椎骨数量较少有关。在种质鉴定方面,岳阳等[9]对7个太湖流域品种与5个西方引进猪种进行简化基因组测序,鉴定并验证了小梅山猪中5个高等位基因突变率的SNPs位点,为小梅山猪种质鉴定提供了科学依据;刘刁等[10]鉴定了深县猪21 602 538个特异性SNPs位点和580个受选择位点,发现其主要参与激素合成、卵细胞成熟、脂质代谢以及免疫等信号通路,为深县猪遗传特性研究提供参考。在分子育种方面,张昌政等[11]利用GWAS和Fst筛选了与体尺性状和乳头数显著关联的SNPs(32和90个),以及与体长、胸围、管围和总乳头数性状显著关联的位点(4个),为苏山猪初生体尺与乳头数性状遗传改良提供了重要的分子标记。
晋汾白猪是由山西农业大学主持培育而成的瘦肉型猪品种,2014年通过国家畜禽遗传资源委员会审定[12]。晋汾白猪将马身猪的抗逆基因、太湖猪(二花脸)的高繁殖特性、长白猪和大白猪的生长速度和瘦肉基因汇聚一身,具有肉质好、产仔多、生长速度快、胴体品质好、肠道微生物种类丰富、抗逆性强等特点[13-14]。晋汾白猪作为培育品种,在长期选育过程中产生了大量的变异位点,如何有效发掘和利用变异位点进行种质特性研究尤为重要。故本研究采用全基因组重测序技术对30头晋汾白猪和18头马身猪进行基因组测序,并整合晋汾白猪亲本重测序数据,鉴定晋汾白猪特异性SNPs并进行特征功能分析,为晋汾白猪种质特性发掘、利用和评价工作提供参考依据。
1 材料与方法
1.1 试验材料
晋汾白猪和马身猪饲养于大同市种猪场,采集晋汾白猪种猪30头(9公、21母)、马身猪18头(11公、7母),剪取面积为1 cm2的耳组织3块,放入1.5 mL离心管内液氮保存。长白猪、大白猪和太湖猪(二花脸)基因组数据从NCBI SRA数据库下载获得(表1)。
表1 长白猪、大白猪和太湖猪(二花脸)基因组数据Table 1 The genome data set of Landrace, Large White and Taihu (Erhualian) pigs
1.2 数据分析软件与仪器设备
本研究中用到的数据分析软件主要包括:minimap2、SAMTOOLS、GATK和DAVID生物信息学网站。用到的主要仪器设备包括:移液器(Eppendorf,德国)、PCR仪(Eppendorf,德国)、高速冷冻离心机(Eppendorf,德国)、电泳仪(六一仪器厂,北京)、水平电泳槽(六一仪器厂,北京)、凝胶成像系统(六一仪器厂,北京)和高压灭菌锅(Sanyo,日本)。
1.3 耳组织DNA提取
取耳组织剪碎置于1.5 mL离心管,加入组织抽提液、蛋白酶K、SDS消化过夜,加入酚-氯仿-异戊醇、无水乙醇进行基因组DNA的提取,后用浓度为1%的琼脂糖凝胶电泳检测DNA纯度和完整性,将合格样品放于-20 ℃保存备用。
1.4 全基因组重测序及比对
1.4.1 DNA文库的构建与测序 经检验合格的DNA用于文库构建和测序,均由康普森公司完成。主要包括Covaris破碎机将DNA随机打碎为350 bp的片段,后经末端修复、加多聚A尾巴、加测序接头、片段纯化、PCR扩增等步骤。质检合格后经Illumina HiSeq对48个样品的DNA片段进行双末端测序,获得的原始reads以fastq格式保存。
1.4.2 测序数据质控及过滤 将测序获得的读段用fastqc软件进行碱基质量和GC含量分析,使用Trimmomatic软件去除接头序列、过滤含N碱基数目大于20%的reads、去除低质量碱基以及reads,获得Clean reads。
1.4.3 序列比对参考基因组 使用minimap2软件(参数:-t 10-K 1G-a-x sr-cs)将Clean reads比对到猪的参考基因组(版本:Sscrofa11.1),并对比对率、测序深度和覆盖度进行统计。
1.5 SNPs位点的检测与注释
将每个样品比对后产生的SAM文件使用SAMTOOLS进行排序并去重复,用GATK软件(v4.0.2)检测变异位点并保存为VCF文件格式,使用VariantRecaliration对每个猪种进行位点重比对,后用VariantFilteration对SNPs位点过滤,得到每个品种SNPs变异位点集合。最后使用VEP软件对其进行注释,统计SNPs的分类和在染色体上的分布。
1.6 特异SNPs位点的筛选
通过对晋汾白猪、大白猪、马身猪、长白猪和太湖猪(二花脸)的SNPs位点进行比较分析筛选晋汾白猪特异SNPs。特异性SNPs位点应满足以下筛选条件:1)等位基因频率(AF)大于0.1;2)仅在晋汾白猪群体中出现的SNPs。提取VEP软件的注释信息,统计在染色体上的分布和分类,将变异位点所在基因利用DAVID数据库进行GO和KEGG富集分析,筛选特异SNPs中分类为错义突变SNPs位点所影响的生物学过程和信号通路。
2 结 果
2.1 基因组样品检测及测序数据质控
本研究对30头晋汾白猪耳组织进行基因组DNA提取。电泳检测 DNA样品条带单一,且OD260 nm/OD280 nm和OD260 nm/OD230 nm在1.88~2.10之间,说明DNA纯度和完整性较好,符合建库和测序要求。
将30份晋汾白猪样品经Illumina平台上机进行全基因组重测序,共获得11 909 696 916条Raw reads(1 786 454 537 400 bp),将低质量的碱基以及reads去除后共得到11 759 401 450条Clean reads(1 761 731 736 206 bp),平均每个样品数据量为58 724 391 207 bp,Q20≥96.1%,Q30≥89.9%,Q30均值为91.77%,GC含量介于42.62%~43.74%之间(表2)。以上结果表明数据质量较高,可用于后续分析。
2.2 重测序数据基因组比对
将过滤获得的Clean reads比对到猪的参考基因组(版本:Sscrofa11.1),由表2可知,30头晋汾白猪基因组测序数据比对率在74.76%~76.59%之间,平均比对率为75.61%,平均覆盖深度为14.7 X,平均覆盖率为96.95%,1 X覆盖度在95.83%以上,5 X覆盖度均值为91.84%,15 X覆盖度均值为48.67%。说明本研究中的30头晋汾白猪基因组DNA测序深度和样品比对率均较高,可用于后续基因组变异检测分析。
2.3 单核苷酸多态性特征分析
将晋汾白猪测序获得的数据与参考基因组进行比对,共检测到99 420 798个SNPs,平均每个样品包含3 314 026.6个SNPs,去冗余后共得到14 680 807个SNPs(8 259 439个SNPs可比对到SNP数据库),转换颠换比(Ti/Tv)平均值为2.55,与之前的结果类似[15-16]。SNPs在猪的每条染色体上分布较均匀且染色体末端分布较多(图1A),其中1号染色体上分布最多(1 341 385个SNPs),13号染色体次之,Y染色体分布最少(50 625个SNPs)(图1B)。以上结果说明,染色体长度越长,分布的SNPs位点越多。

图1 晋汾白猪SNPs的染色体密度(A)、数目(B)和类型(C)Fig.1 Density (A), number (B) and type (C) of SNPs loci on chromosomes in Jinfen White pigs
对SNPs的类型进行统计,有708 428(4.83%)和741 843(5.05%)个SNPs分别位于基因上、下游,308 103个SNPs(2.10%)位于外显子区,内含子区SNPs变异最多,有7 194 452 个(49.01%),其次位于基因间隔区(37.53%),剪接位点区域内的SNPs数目最少,有22 028个(图1C)。
2.4 晋汾白猪特异SNPs位点的筛选
为了筛选晋汾白猪特异性SNPs位点,本研究随后对18头马身猪基因组进行重测序, 并在NCBI的SRA数据库下载30头长白猪、17头大白猪、40头太湖猪(二花脸)的基因组重测序数据(表1,表3)。对以上5个品种猪基因组重测序数据进行统计分析,共测到5 772.84 Gb的原始数据,经过质控和过滤后共得到5 584.50 Gb的纯净数据,共有31 560 947 644条reads可以比对到参考基因组(表3)。
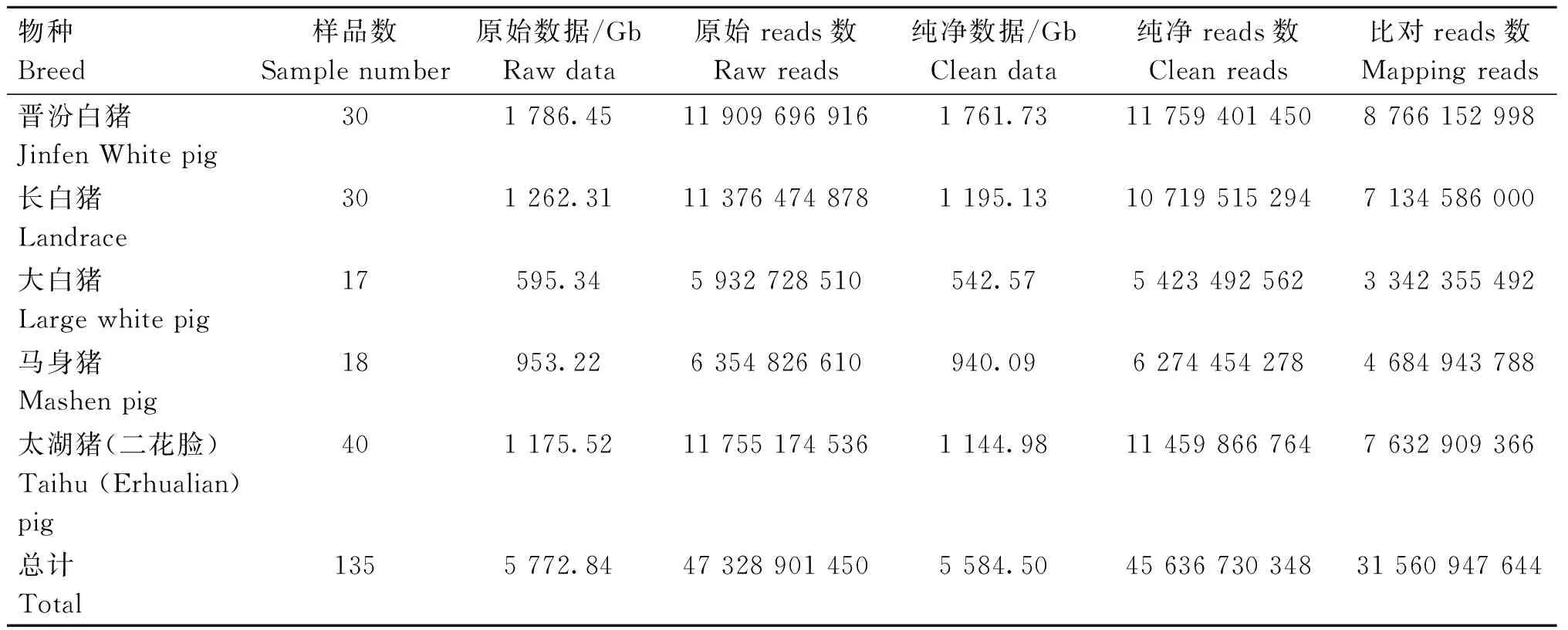
表3 5个品种猪重测序数据统计Table 3 Summary of resequencing data in 5 different pig breeds
将30头晋汾白猪样品作为一个群体,其余品种作为另一个群体,基于等位基因频率(AF>0.1)筛选晋汾白猪特异SNPs,共获得了87 366个特异的SNPs位点,平均每28.44 kb就有1个特异SNP(猪基因组总长2.44 Gb)(图2A)。其中2号染色体上分布最多(8 650个SNPs),1号和15号染色体次之(7 477和7 267个SNPs),Y染色体分布最少,仅有530个,且这些特异性的SNPs分布于25 606个基因上(图2B)。对晋汾白猪特异SNPs进行分类,发现这些特异性的SNPs类型占比与晋汾白猪所有SNPs的类型占比相似,主要分布在内含子区(49.50%)和基因间区(37.58%),剪接位点和5′非翻译区最少,均不到1%(图2C)。

图2 晋汾白猪特异性SNPs的染色体密度(A)、数目(B)和类型(C)Fig.2 Density (A), number (B) and type (C) of specific SNPs loci on chromosomes in Jinfen White pigs
2.5 特异SNPs功能富集分析
有993个特异性SNPs位点位于外显子区,其中包括555个同义突变SNPs,437个错义突变SNPs和1个无义突变SNPs,这些错义突变SNPs位点分布在343个基因上,包括影响猪肌肉生长发育的基因ITGA4、MYH和ANK1,影响免疫应答的基因FN1、DNER和ITSN2等。对以上343个基因进行GO富集分析,共富集到245个条目,包括生物学过程(137)、分子功能(51)和细胞组分(57),其中包括17个显著富集的条目(P<0.05),细胞组分富集的条目最多(12个)。大部分基因(gene number>15)富集在胞内无膜细胞器(intracellular non-membrane-bounded organelle)、无膜细胞器(non-membrane-bounded organelle)、退化的细胞器组分(absolete organelle part)、退化的胞内细胞器组分(absolete intracellular organelle part)等过程(图3A)。此外,对以上343个基因进行KEGG富集分析,共富集到178个信号通路,显著富集到9个信号通路(P<0.05),包括肌动蛋白细胞骨架调节(regulation of actin cytoskeleton)、初级胆汁酸生物合成(primary bile acid biosynthesis)、肠道免疫系统IgA的合成(intestinal immune network for IgA production)、过氧化物酶(peroxisome)、粘着斑(focal adhesion)、磷脂酶D信号通路(phospholipase D signaling pathway)、脂肪酸降解(fatty acid degradation)、轴突导向(axon guidance)、急性髓细胞白血病(acute myeloid leukemia),其中肌动蛋白细胞骨架的调节信号通路富集的基因最多(8个),脂肪酸降解和初级胆汁酸生物合成通路只富集到了2个基因(图3B)。通过以上富集分析,发现IGTA4、SLC27A2、MYH、PIK3R、PDFGA、FN1、SSH、CEBPE、PAK3、MHC2、ROBO3、APC5、MAP3K14、CSF1R等富集于上述信号转导、生长发育、肠道免疫、脂质代谢等过程,以上基因的特异性SNPs位点的鉴定将为晋汾白猪种质特异性研究提供参考依据。

图3 晋汾白猪特异性错义突变SNPs位点所在基因的GO(A)和KEGG(B)富集分析(P<0.05)Fig.3 GO (A) and KEGG (B) enrichment analyses of specific missense mutation SNPs loci in Jinfen White pigs (P<0.05)
3 讨 论
近年来,以二代测序技术为代表的全基因组测序推动了畜禽动物基因组的注释以及分析工作,与从头测序不同,全基因组重测序(resequencing)是指对已知物种个体基因组再次测序,并同参考基因组进行比对分析,获得遗传变异信息,在个体和群体差异比较、进化研究、重要经济性状候选基因筛选等方面起到了重要的作用[17]。Jiang等[18]通过对无后肢突变猪、亲代母猪和同代正常仔猪进行全基因组重测序并进行比较基因组学分析,筛选出6个可能导致表型变异的候选基因(包括骨形态发生蛋白BMP7),为解析肢体表型变异原因提供了参考。此外,Miller等[19]对1只大角羊进行三代基因组重测序并与其他绵羊基因组比对,共发现1 562万个遗传变异,其中同义和非同义突变SNPs所在基因主要富集于精子发生和乳腺上皮细胞增殖负调控等信号通路,为绵羊遗传机制的解析和育种奠定了理论基础。Herrero-Medrano等[20]对欧洲猪种进行重测序,鉴定了99个同义突变SNPs(65个基因),并筛选AZGP1和TAS2R40作为环境适应性候选基因。Liu等[21]对清平猪和其他品种猪全基因组重测序数据进行整合,筛选了10 342 544个高质量的SNPs位点并结合RNA-Seq数据,鉴定出EGFR可能与猪的妊娠期长短有关。
测序质量的好坏通常用测序深度和覆盖度来衡量[22]。测序深度反映测序量的大小,与结果的准确性成正比,一般的测序深度可达到10 X~30 X,且覆盖度与测序深度呈一致性。李文婷[23]对6个猪种进行重测序,平均测序深度和覆盖率分别为10 X和 94%。Choi等[24]对韩国本土猪、野猪和欧洲猪种进行基因组重测序,平均测序深度和覆盖率高达11.7 X和 99.2%。本研究共得到约1 786.45 Gb的重测序数据,Q30高达89.9%,平均测序深度高达14.7 X,且平均覆盖率为96.95%,参考基因组平均比对率为75.61%。说明晋汾白猪基因组重测序与其他猪种研究结果类似,具有数据大、覆盖率高、质量和比对情况良好等特征,为后续数据分析提供了可靠的保障。
通过去冗余共鉴定了14 680 807个SNPs位点,主要分布于1号染色体,Y染色体分布最少(0.34%)。与崔清明等[25]利用RAD-Seq简化基因组测序结果相似,在480份大围子猪保种核心群中鉴定了387 935个SNPs位点,其中有27 906个分布于1号染色体(7.17%),仅有1 143个分布于Y染色体(0.19%),说明染色体上SNPs的分布可能与染色体的长度有关。此外,本研究大部分SNPs分布在内含子区和基因间隔区,外显子区和剪接位点分布的最少。与任钰为等[26]的研究结果类似,五指山猪和杜洛克猪的基因组重测序结果中大部分SNPs分布于内含子区域和基因间区,而编码区的SNPs则较少。以上结果说明,与多数研究结果类似,编码区的SNPs少于非编码区,这可能与基因组的非编码区占比高有关,也可能与其分布在启动子区或者非编码区发挥转录调控功能有关[26-27]。
通过将晋汾白猪的SNPs与马身猪、长白猪、大白猪、太湖猪(二花脸)的SNPs进行特异性比较分析,基于等位基因频率共鉴定了87 366个(0.6%)晋汾白猪特异性SNPs位点。这些位点主要分布于25 606个基因上,覆盖了基因组大部分区域,且多数位于2号染色体。李文婷[23]利用6个品种共48头猪全基因组重测序数据,获得了132 078个太湖猪特有的SNPs位点,高于本研究中筛选的晋汾白猪特异性SNPs位点,可能是由于品种遗传多样性不同而产生的差异。
错义突变将对编码蛋白质产生影响,可能直接或者间接造成品种间性状差异。因此,对包含有错义突变SNPs位点的343个基因进行GO和KEGG功能富集分析,发现主要影响细胞器和细胞器组分等生物学过程,且部分基因显著富集于肌动蛋白细胞骨架调节、初级胆汁酸生物合成、肠道免疫系统IgA的产生、过氧化物酶、磷脂酶D、脂肪酸降解、急性髓细胞白血病等过程,说明这些SNPs位点可能影响细胞生长、骨骼肌生长发育、机体代谢和免疫等信号通路。在以上所有富集的生物学过程(245个)和信号通路(178个)中包括影响机体发育的基因MYH(蛋白结合、肌动蛋白细胞骨架调节、血管平滑肌收缩)和ANK1(细胞质、细胞膜、代谢途径),影响免疫应答的基因FN1(肌动蛋白骨架调节、黏着斑信号通路、ECM受体互作、癌症蛋白聚糖等)、DNER(细胞膜)和ITSN2(结合)。MYH作为肌球蛋白重链的组成部分不仅可以影响骨骼生长发育而且可以调控肌纤维类型,进而改善猪肉品质[28-30]。Lee等[31]对181头杜洛克猪和14个肉品质相关性状进行全基因组关联分析,发现5个基因上的SNPs位点与肉品质相关,其中就包括ANK1。此外,ANKI作为转录因子在造血干细胞向髓系祖细胞分化过程中表达量上调并发挥作用[32]。Xie等[33]为了研究miR-183对脂多糖诱导的断奶仔猪海马体氧化应激的调控作用,通过转染miR-183 mimic以及利用荧光素酶报告基因等试验,发现miR-183可通过靶向FN1调节脂多糖诱导的氧化应激。Wang等[34]发现,Delta和Notch样内皮生长因子相关受体DNER,可作为跨膜蛋白传递神经细胞和胶质细胞间的信号,通过抑制TOR4A表达和功能影响胶质瘤产生。此外,ITSN2作为一种多器官高度保守的支架蛋白,主要参与T细胞活性和卵母细胞发育等过程[35-36]。在鸡骨骼肌发育过程中,ITSN2转录产生的环状RNA circITSN2可充当分子海绵结合miR-218-5p增加LMO7的表达促进鸡胚胎成肌细胞的增殖和分化[37]。以上结果说明,晋汾白猪可能在肌肉生长发育和免疫应答方面有别于亲本,但又继承了亲本的优点。在后续的工作中,将对特异性SNPs进一步筛选,用于分子标记开发基因组选种芯片检测试剂盒,以及晋汾白猪纯种鉴定和种质资源采集。
4 结 论
本研究通过对30头晋汾白猪基因组重测序,共得到了14 680 807个SNPs位点,基于等位基因频率剔除与亲本相关的变异位点,筛选到87 366个晋汾白猪特异SNPs,包括影响机体发育的基因MYH和ANK1和影响免疫应答的基因FN1、DNER和ITSN2等。本研究结果从基因组水平上解析了晋汾白猪种质特征形成基础,为晋汾白猪选育、纯种鉴定和核心种质的筛选提供了理论依据。
