肠道/口腔微生物组、血液代谢物等与胃癌相关性
2023-07-26穆雪峰李振宇黄书琦康文全尹小菲穆桂芳
穆雪峰 李振宇 黄书琦 康文全 尹小菲 穆桂芳
(1华中科技大学协和深圳医院消化科,广东 深圳 518052;2深圳微辰生命科技有限公司;3威海市立二院消化科)
胃癌发病率居恶性肿瘤第5位,死亡率居第4位,总体死亡率为7.7/10万。近5年全球胃癌平均年发病例数为180.6万例,其中亚洲139.7万例(77.4%),中国68.9 万例(38.2%)。我国 2020 年胃癌发病率居恶性肿瘤第 3 位,死亡率居第 3 位,死亡37.4万例,死亡率15.9/10万〔1〕。统计结果显示,胃癌在老年人群中高发,发病高峰为65~80岁,80岁以上胃癌患者死亡率最高。幽门螺旋杆菌通常被认为是导致胃癌的重要因素〔1〕。研究表明,除幽门螺旋杆菌,其他微生物也可能在胃癌的发生、发展过程中发挥重要的作用〔2,3〕,同时血液中相关代谢物也和胃癌发病及进展存在密切关系〔4~6〕。本文收集粪便、唾液和血液样本进行研究,以期得到和胃癌相关的潜在生物标志。
1 材料与方法
1.1对象 纳入华中科技大学协和深圳医院胃癌患者5例为研究组,选取同期正常且无其他病史的人群5例作为对照组。性别、年龄和胃癌患者尽量匹配。对所有招募者进行问卷调查,主要包括年龄、民族、性别、吸烟和饮酒史、既往慢性基础病病史及相关辅助检查等特征资料。同时检测胃癌相关临床指标,建立临床信息数据库,如血常规,胃癌的肿瘤标记物癌胚抗原(CEA)、糖类抗原(CA)19-9、CA125等。采集样本包括血液、粪便、唾液。纳入标准:研究组采用病理活检、胃镜等诊断,确诊疾病;资料齐全;认知正常;熟知本研究,自愿参加。排除标准:精神异常;其他恶性肿瘤;胃大部切除术;依从性差。两组年龄等一般资料差异无统计学意义(P>0.05)。
1.2样本采集 粪便和唾液样本均在未接受任何治疗的情况下采集。粪便样本采集:采集前受试者需先排尿,然后将粪便排于无菌采集袋中,排便结束后用一次性采样勺采集中间未接触空气部分0.5~0.8 g样本,将样本置于冻存管中拧紧后-80 ℃冻存。唾液样本采集:采集前30 min受试者需先用清水漱口,用带有漏斗的采集管收集唾液约2 ml,取下采集漏斗并拧紧采集管后-80 ℃冻存。
1.3基因组DNA提取和建库测序 使用Omega E.Z.N.A.Stool DNA Kit (Bio-tek,Inc.,USA) 提取粪便和唾液的微生物基因组DNA。提取完成的DNA用Covaris E220超声打断仪打断成约300 bp的小片段后,使用NEB Next Ultra DNA Library Prep Kit进行测序文库构建,构建好的文库经过Qubit 定量和文库检测合格后,在Illumina Novaseq6000平台进行PE150的文库测序,并保证每个样本下机数据量不低于5G。
1.4统计学处理 测序得到的下机数据的处理步骤:(1)过滤低质量的数据;(2)使用SOAP2比对到基因集;(3)根据基因集和微生物分类及KO的对应关系,计算门(phylum)、属(genus)、种(species)和KO的相对丰度〔7〕。样本内的物种多样性(Alpha多样性)使用shannon index表示。样本间的物种多样性(Beta多样性)使用bray-curtis距离表示。主坐标分析(PCoA)用来展示样本间的bray-curtis距离。相似性分析(ANOSIM,置换次数为999)用来评估不同组的样本的分布差异是否有统计学意义。使用R(版本3.5.1)中的wilcox秩和检验用于对不同组之间的微生物的相对丰度进行差异分析。肠型分析:(1)基于样本在属水平的相对丰度计算样本间JSD;(2)基于JSD进行围绕中心点划分算法(PAM)聚类;(3)使用CH指数评估最优聚类数量;(4)使用PCoA展示聚类效果〔8〕。线性判别分析(LEfSe)用于对不同组样本的微生物相对丰度进行比较,以获得可能和胃癌相关的微生物〔9〕。LDA分值的阈值设定为3。使用R(版本3.5.1)中的cor.test在属水平对微生物的相关性进行分析。选择相关系数<-0.6或>0.6,P值<0.05,且在不同组的样本中的相对丰度之和>0.1%的微生物使用cytoscape进行绘图展示。随机森林模型用于对不同组的样本进行分类,最终的曲线下面积(AUC)为5折交叉验证的均值。基于样本的KO相对丰度,使用reporter score算法计算KEGG模块和通路的z-score〔10〕。z-score的绝对值>1.96(正态分布的97.5%置信度)时,认为结果在不同组之间有显著差异。使用R(版本3.5.1)psych包中的corr.test对临床指标和LEfSe找到的差异微生物进行关联分析。检验方法为spearman。
2 结 果
2.1Alpha和Beta多样性 研究组和对照组肠道微生物和口腔微生物在属和种水平的Alpha多样性shannon差异均无统计学意义(P>0.05)。但研究组肠道微生物和口腔微生物的Alpha多样性的中位数均低于对照组。见图1。研究组和对照组在PCoA1和PCoA2维度的分布无法明显区分。ANOSIM分析的结果表明研究组和对照组差异无统计学意义。口腔微生物的结果也同样如此(P>0.05)。见图2。

A:肠道微生物,属水平;B:肠道微生物,种水平;C:口腔微生物:属水平;D:口腔微生物,种水平;图2同图1 两组肠道和口腔微生物的Alpha多样性

图2 两组肠道和口腔微生物的Beta多样性
2.2微生物构成和肠型 在门水平,相对丰度较高的微生物包括拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)、放线菌门(Actinobacteria)等。在属水平,相对丰度较高的微生物包括拟杆菌属(Bacterodie)、经黏液真杆菌属(Blautia)、普雷沃菌属(Prevotella)、普拉梭菌(Bifidobacterium)、大肠杆菌(Escherichia)等。其中,研究组和对照组中至少有一组的相对丰度中位数大于0.1%,两组差异具有统计学意义,乳杆菌属内(Lactobacillus)(研究组1.282 6%,对照组0.001 6%,P=0.016)。在种水平,相对丰度较高的微生物包括Bacteroides dorei、Bacteroides uniformis、Bacteroides ovatus等。其中,两组中至少有一组的相对丰度的中位数大于0.01%,差异具有统计学意义;咽颊炎链球菌(Streptococcus anginosus)(研究组0.047%,对照组0.001%,P=0.008)、粪球菌属(Coprococcus comes)(研究组0.078%,对照组=0.010%,P=0.016)、克雷伯杆菌(Klebsiella pneumoniae)(研究组0.154%,对照组0.02%,P=0.016)、Lactobacillus salivarius(研究组0.017%,对照组0.000%,P=0.016)、Dorea longicatena(研究组0.054%,对照组0.006%,P=0.032)、布氏瘤胃球菌(Ruminococcus bromii)(研究组0.325%,对照组0.003%,P=0.032)。
研究组和对照组中丰度较高的口腔微生物,在门水平,相对丰度较高的微生物包括Proteobacteria、Firmicutes、Bacteroidetes、梭杆菌门(Fusobacteria)、Actinobacteria等。在属水平,相对丰度较高的微生物包括嗜血杆菌属(Haemophilus)、链球菌属(Streptococcus)、韦荣球菌属(Veillonella)、伯克霍尔德菌属(Burkholderia)、Fusobacterium等。研究组和对照组中至少有一组相对丰度的中位数大于0.1%,且在两组差异具有统计学意义,包括Actinobacillus(GCmedian=0.007%,Control median=0.106 %,P=0.032)。在种水平,相对丰度较高的微生物包括副流感嗜血杆菌(Haemophilus para influenzae)、炎鼻疽伯克霍尔德菌(Burkholderia pseudomallei)、未分类的乳杆菌(unclassified Fusobacterium)、弯曲杆菌(Campylobacter concisus)等。两组中至少有一组的相对丰度的中位数大于0.01%,且在两组之间的差异具有统计学意义,包括Actinobacillus minor(研究组0.00%,对照组0.01%,P=0.011)、Actinobacillus pleuropneumoniae(研究组0.000%,对照组0.016%,P=0.025)、Kingella denitrificans(研究组0.003%,对照组0.018%,P=0.032)、Neisseria mucosa(研究组0.025%,对照组0.164%,P=0.032)、Streptococcus mutans(研究组0.005%,对照组=0.016%,P=0.032)。见图3。

A:肠道微生物,门水平;B:肠道微生物,属水平;C:肠道微生物,种水平;D:口腔微生物,门水平;E:口腔微生物,属水平;F:口腔微生物:种水平图3 两组相对丰度较高的肠道微生物和口腔微生物
研究组和对照组的所有肠道样本可以聚为2类,分别对应肠型1和肠型2。见图4。研究组均属于肠型1。对照组中4例肠型1,1例肠型2。两组在不同肠型中的人数分布的差异不具有统计学意义(fisher检验,P=1)。
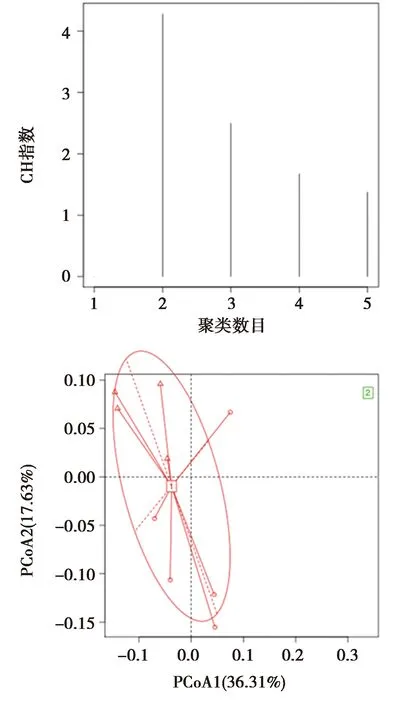
图4 两组粪便样本的肠型分析结果
2.3与胃癌相关的微生物及分类模型 在门、属和种水平进行了LEfSe来寻找可能和胃癌相关的微生物。在肠道微生物中共找到7种分类,包括1种门水平的分类,2种属水平的分类、4种种水平的分类见图5A。在门水平,Firmicutes在研究组中的相对丰度较高。在属水平,在研究组中的相对丰度较高的Lactobacillus属于Firmicutes门,Thauera属于Proteobacteria;和相关研究一致〔11~14〕。在种水平,研究组中的相对丰度较高的4种分类中,Lactobacillus reuteri属于Firmicutes门下Lactobacillus属。
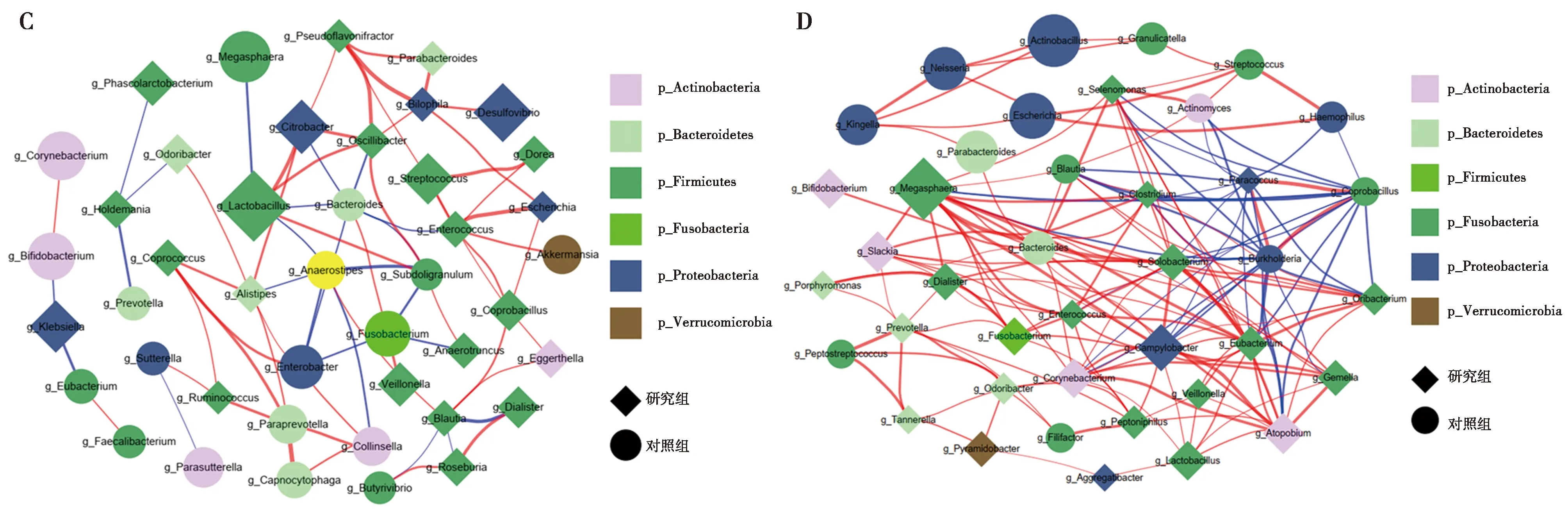
A:可能和胃癌相关的肠道微生物;B:可能和胃癌相关的口腔微生物;C:肠道微生物之间相关性网络;D:口腔微生物之间相关性网络;相关性网络中,节点大小正比于研究组和对照组之间微生物的相对丰度中位数之比;边的粗细正比于相关系数的大小的绝对值,蓝色边表示负相关,红色边表示正相关图5 LEfSe分析获得的可能和胃癌相关的微生物及微生物之间的相关性
在口腔微生物中共找到6种分类和胃癌相关,包括3种属水平的分类、3种种水平的分类见图5B。在属水平,2种在研究组中的相对丰度较高的属中,Campylobacter属于Proteobacteria门;Gemella属于Firmicutes门。在对照组中的相对丰度较高的Neisseria属于Proteobacteria门;和之前报道一致〔15~17〕。在种水平,对照组中的相对丰度较高的3种分类中,Campylobacter concisus 属于Proteobacteria门的Campylobacter属; Jonquetella anthropi属于Synergistetes门的Jonquetella属;Gemella sanguinis属于Firmicutes门的Gemella属。
基于微生物之间的相关性分析,获得部分相关性较高且具有一定相对丰度的微生物。肠道微生物中,和Lactobacillus正相关的属包括Citrobacter (r=0.85,P=0.003)、Oscillibacter(r=0.79,P=0.01)等。在对照组中相对丰度更高的分类包括Bifidobacterium、Eubacterium、Faecalibacterium、Subdoligranulum等有益菌。这些有益菌能够生成丁酸盐、丙酸盐、醋酸盐等对人体健康有益的代谢物〔18~21〕。见图5C。
口腔微生物中,Campylobacter和Gemella之间的相关性较高(r=0.83,P=0.006)。和Campylobacter相关性较高的其他属包括Atopobium (r=0.93,P=0.000 1)、Megasphaera(r=0.89,P=0.001)、Solobacterium(r=0.88,P=0.002)等。和Gemella相关性较高的其他属包括Atopobium(r=0.88,P=0.002)、Corynebacterium(r=0.82,P=0.007)等。和Neisseria相关性较高的其他属包括Kingella(r=0.88,P=0.002)、Actinobacillus(r=0.79,P=0.01)等,见图5D。
在肠道微生物和口腔微生物中,研究组Lactobacillus、Enterococcus、Veillonella、Dialister、Odoribacter相对丰度均大于对照组;Bacteroides相对丰度均小于对照组。
基于LEfSe找到的胃癌相关的微生物,使用随机森林模型构建分类模型 。在肠道微生物中,分别使用属水平的Lactobacillus和陶厄菌属(Thauera)中的一种作为特征时,AUC分别为0.9、0.7;当同时使用这些属作为特征时,AUC为0.9。在口腔微生物中,分别使用属水平的Campylobacter、Gemella和Neisseria中的一种作为特征时,AUC分别为0.7、0.9、0.8;当同时使用这些属作为特征时,AUC为0.8。见图6。
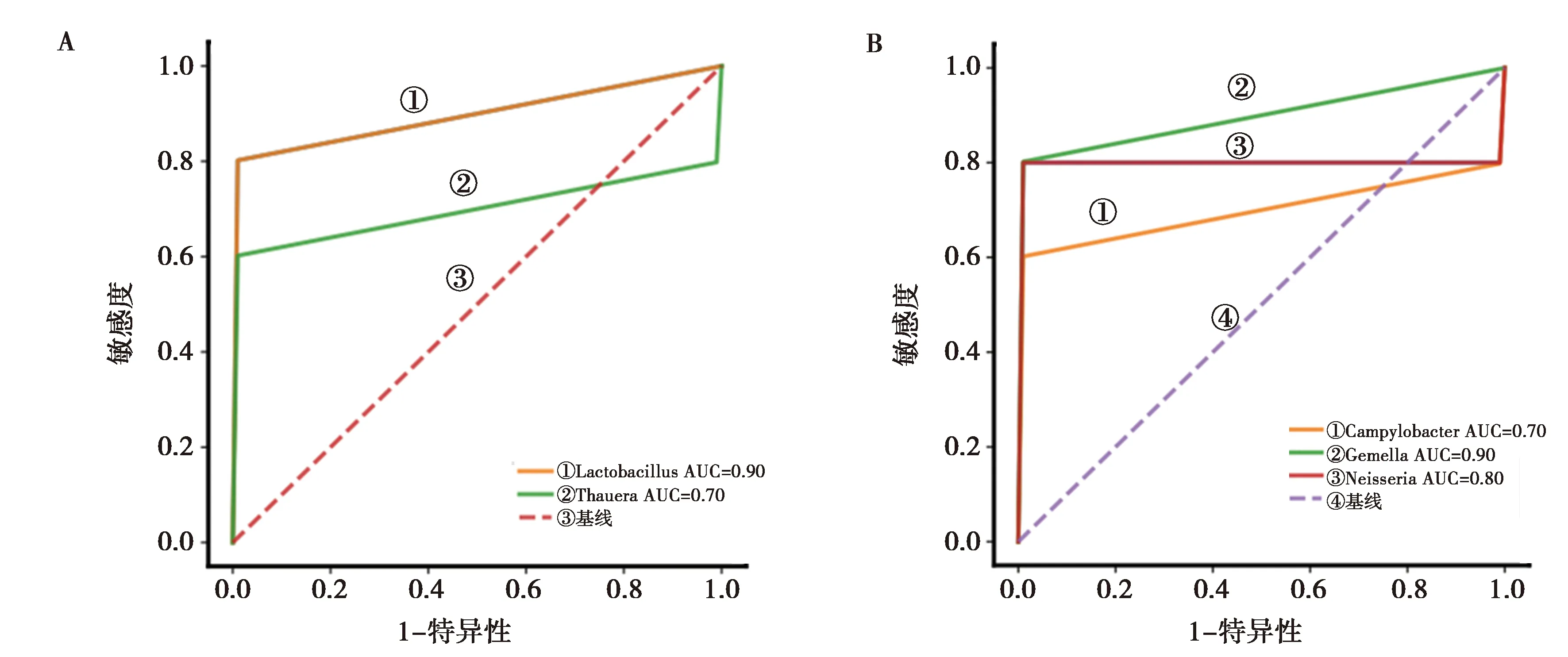
A:肠道微生物;B:口腔微生物;图7、8同图6 基于可能和胃癌相关的微生物构建的分类模型的受试者工作特征(ROC)曲线
2.4和胃癌相关的临床指标及微生物关联分析 对检测的10个临床指标进行差异分析。其中CEA和鳞状细胞癌相关抗原(SCCa)在两组差异不具有统计学意义(wilcox秩和检验,CEA:研究组8.66,对照组2.10,P=0.06;SCCa:研究组0.27,对照组0.71,P=0.06)。
计算临床指标的检测值和LEfSe找到的和胃癌相关的微生物分类之间的相关性。在粪便样本中,CEA的检测值和属水平的Thauera的相对丰度呈正相关(r=0.76,P=0.01)。SCCa的检测值和属水平的Lactobacillus、种水平的Lactobacillus reuteri、Alistipes putredinis、Streptococcus_anginosus的丰度呈负相关(r分别为-0.85、-0.79、-0.87、-0.68,P分别为0.001、0.006、0.001、0.030)。见图7。
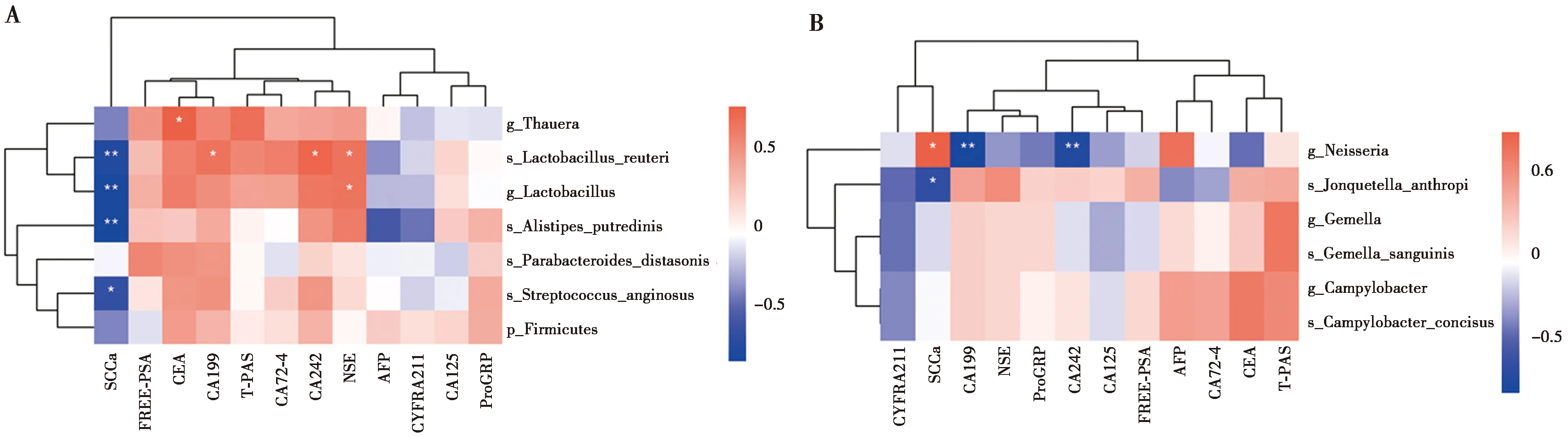
图7 临床指标和微生物之间的关联性
在唾液样本中,SCCa和属水平的Neisseria的丰度呈正相关(r=0.72,P=0.02)。CA242、CA199和Neisseria的丰度呈负相关(r分别为-0.77、-0.84,P分别为0.009、0.002)。见图7。
2.5功能分析 基于reporter score算法的富集分析,获得在分别研究组和对照组中富集的KEGG通路。肠道微生物中,两组之间显著富集的通路明显不同。 研究组显著富集的通路主要和遗传信息处理、脂质代谢等相关。而在对照组中显著富集的通路主要和氨基酸代谢、辅助因子和维生素代谢等相关。研究组显著富集的通路主要和遗传信息处理、脂质代谢等相关。见图8。


--:reporter score<-2.3,-:reporter score<-1.96,++:reporter score>2.3,+:reporter score>1.96图8 两组富集的KEGG通路
3 讨 论
从物种构成来看,肠道微生物和口腔微生物的相对丰度最高的3种门都是Bacteroidetes、Firmicutes和Proteobacteria。但是肠道微生物中相对丰度最高的Bacteroidetes在口腔微生物中的相对丰度较低。而肠道微生物中相对丰度较低的Proteobacteria在口腔微生物中的相对丰度则最高。
LEfSe发现一些和胃癌相关的微生物。尽管在属水平,不同样本类型(粪便、唾液)发现的相关微生物不一样,但在门的层级存在共性。例如在研究组中相对丰度更高的肠道微生物中的Lactobacillus和口腔微生物中的Gemella都属于Firmicutes门。当使用和胃癌相关的微生物构建分类模型时,肠道微生物中发现的Lactobacillus和口腔微生物中发现的Gemella的分类效果相当。通过相关性分析发现,肠道微生物中的Enterococcus、Dorea和Streptococcus等在研究组的相对丰度大于对照组,而这些微生物也曾在之前的研究中被认为和胃癌相关。说明不同的样本情况(包括数量、临床特征等)、检测方法(16S、宏基因组测序等)及分析方法等均可能影响结果。相关性分析发现明肠道微生物和口腔微生物之间存在一定一致性。
肠道微生物中,翻译(RNA转运、核糖体)、转录(RNA聚合酶)通路在研究组中显著富集,这些通路和癌症的发生、发展密切相关〔22,23〕。磷酸转移酶系统和ABC运送等膜转运通路也在对照组中显著富集。Firmicutes门是对这些通路做出贡献的主要微生物之一〔24〕。脂质代谢(甘油脂代谢、甘油磷脂代谢、酮体合成和降解)、异物生物降解与代谢(苯酸盐降解、氟苯甲酸酯降解、多环芳烃降解)。Liao等〔25〕研究发现多环芳烃的一种代谢产物1-羟基芘葡萄糖醛酸苷在胃癌患者中的浓度要比健康对照的高。与此相反,氨基酸代谢(丙氨酸-天冬氨酸和谷氨酸代谢、缬氨酸-亮氨酸和异亮氨酸生物合成、苯丙氨酸-酪氨酸和色氨酸生物合成、组氨酸代谢)、能量代谢(氧化磷酸化、原核生物中的碳固定通路)、辅助因子和维生素代谢(卟啉和叶绿素代谢、生物素代谢、叶酸合成、维生素B6代谢)等通路则在对照组中显著富集。维生素对于维持健康状态发挥着重要的作用〔26〕。和维生素B6合成呈负相关的Blautia、Roseburia在健康对照中的相对丰度更低,和生物素合成相关的Bacteroides则在对照组的丰度更高〔8,27〕。口腔微生物中,异物生物降解和代谢(苯酸盐降解、氯环己烷和氯苯降解、多环芳烃降解)、神经退行性疾病(阿尔茨海默病、帕金森病)等通路在胃癌患者中显著富集。
综上,胃癌患者组和健康人无论在肠道微生物还是口腔微生物方面都存在差异。这些差异既存在共性,也存在特性。肠道微生物中的Lactobacillus可以作为一种潜在的和胃癌相关的生物标记。由于本研究中样本数量的限制,分析存在一定局限性,后续使用更大样本量进行分析可以进一步完善现有结果。
