儿童原发性扩张型心肌病的临床特征及遗传学分析
2023-07-17郑奎武菲娄美娜王莹雪李博郝京霞王永丽张英谦齐焕军
郑奎 武菲 娄美娜 王莹雪 李博 郝京霞 王永丽 张英谦 齐焕军
(1.河北医科大学研究生学院,河北石家庄 050017;2.河北省儿童医院心内科/河北省小儿心血管重点实验室,河北石家庄 050031)
扩张型心肌病(dilated cardiomyopathy,DCM)是儿童心肌病中最常见的类型,也是导致儿童心源性猝死(sudden cardiac death,SCD)和心力衰竭的常见原因之一[1-2]。DCM病因复杂,不同病因与患儿的预后显著相关[2]。当前儿童DCM 的诊疗虽然取得了一定进展,但早期精准化病因诊断困难、病死率高等问题仍没得到显著改善,且缺乏有效的危险分层方案。随着基因检测技术的发展及应用,30%~40%的DCM患儿具有遗传学病因基础[3],并且多项研究显示存在致病基因突变的DCM患儿病死率更高[1,4]。不同突变基因型、同一基因不同突变位点及环境因素等均与患儿预后密切相关[5]。进一步深入研究儿童DCM基因型-表型的关系有利于患儿的精准预后评估及治疗。当前国内关于儿童DCM 遗传学病因研究报道较少,且缺乏根据遗传学背景来精准评估患儿预后的临床资料[6]。本研究通过对比基因突变阳性和基因突变阴性DCM 患儿之间的临床特征及预后,并分析基因突变阳性患儿的遗传学特征,以期为DCM 患儿的精准预后评估提供依据。
1 资料与方法
1.1 研究对象
回顾性选取2018年7月—2023年2月河北省儿童医院收治的44 例原发性DCM 患儿为研究对象,且所有患儿均已行基因检测。DCM 诊断标准参考2019 年《AHA 儿童心肌病的分类和诊断科学声明解读》[1],排除炎症性、高血压、心脏瓣膜病、缺血性心脏病、先天性心脏病、心动过速、化疗药物或维生素D 缺乏等导致的继发性DCM。所有患儿均接受常规抗心力衰竭药物治疗(米力农、地高辛、血管紧张素转化酶抑制剂、利尿剂等),定期于我院心内科门诊规律复诊。
根据基因检测结果将44 例患儿分为基因突变阳性组(17例)和基因突变阴性组(27例)。本研究获得患儿监护人知情同意,通过河北省儿童医院伦理委员会审批(202136号)。
1.2 基因检测
基因检测均选用全外显子组测序技术(不含线粒体基因),委托第三方公司(北京迈基诺基因科技股份有限公司医学检验所或福州福瑞医学检验实验室有限公司)完成检测,所有患儿家属行Sanger 测序进行验证。根据2015 年美国医学遗传学与基因组学学会指南[7]对变异的致病性进行分类。实验室报告的致病变异或疑似致病变异定义为基因突变阳性;未检出明确与临床表型相关的致病/疑似致病变异,或检出临床意义未明变异但与患儿临床表型无关或家族其他成员无心肌病表型(不符合家系遗传模式)的定义为基因突变阴性。
1.3 资料采集
临床资料主要通过查阅电子病历系统收集,包括首诊时年龄、性别、临床表现(主诉及体征)、既往史、家族史(心肌病或SCD)、实验室检查、心电图、动态心电图及超声心动图等。
1.4 统计学分析
采用SPSS 25.0 统计学软件对数据进行分析。计数资料以例数和百分率(%)表示,组间比较采用卡方检验或Fisher确切概率法。符合正态分布的计量资料以均数±标准差(±s)表示,组间比较采用两样本t检验;偏态分布的计量资料以中位数(四分位数间距)[M(P25,P75)]表示,组间比较采用Mann-WhitneyU检验。以P<0.05 为差异有统计学意义。
2 结果
2.1 两组首诊时临床资料比较
44 例患儿中,男性21 例(48%),女性23 例(52%),中位首诊年龄为12(7,96)个月;首诊临床表现以咳嗽、气促等呼吸道症状最常见(34%,15/44),其次以食欲差、腹痛、呕吐等胃肠道症状多见(27%,12/44)。两组患儿性别、家族史、首诊年龄、首诊时临床表现、心功能分级Ⅲ~Ⅳ级比例,以及首诊时脑钠肽、肌钙蛋白Ⅰ、肌酸激酶同工酶等比较差异均无统计学意义(P>0.05),见表1。
2.2 两组首诊时心电图、超声心动图检查结果比较
患儿首诊时心电图以ST-T 段改变最常见,占73%(32/44),其次分别以窦性心动过速(34%,15/44)、室性期前收缩(27%,12/44)、左室高电压(27%,12/44)较为常见。基因突变阳性组患儿首诊时心电图表现为PR 间期延长、左室高电压、窦性心动过速比例高于基因突变阴性组(P<0.05)。两组患儿首诊时左心室射血分数(left ventricular ejection fraction,LVEF)和左心室短轴缩短率(left ventricular fraction shortening,LVFS)差异均无统计学意义(P>0.05)。见表2。
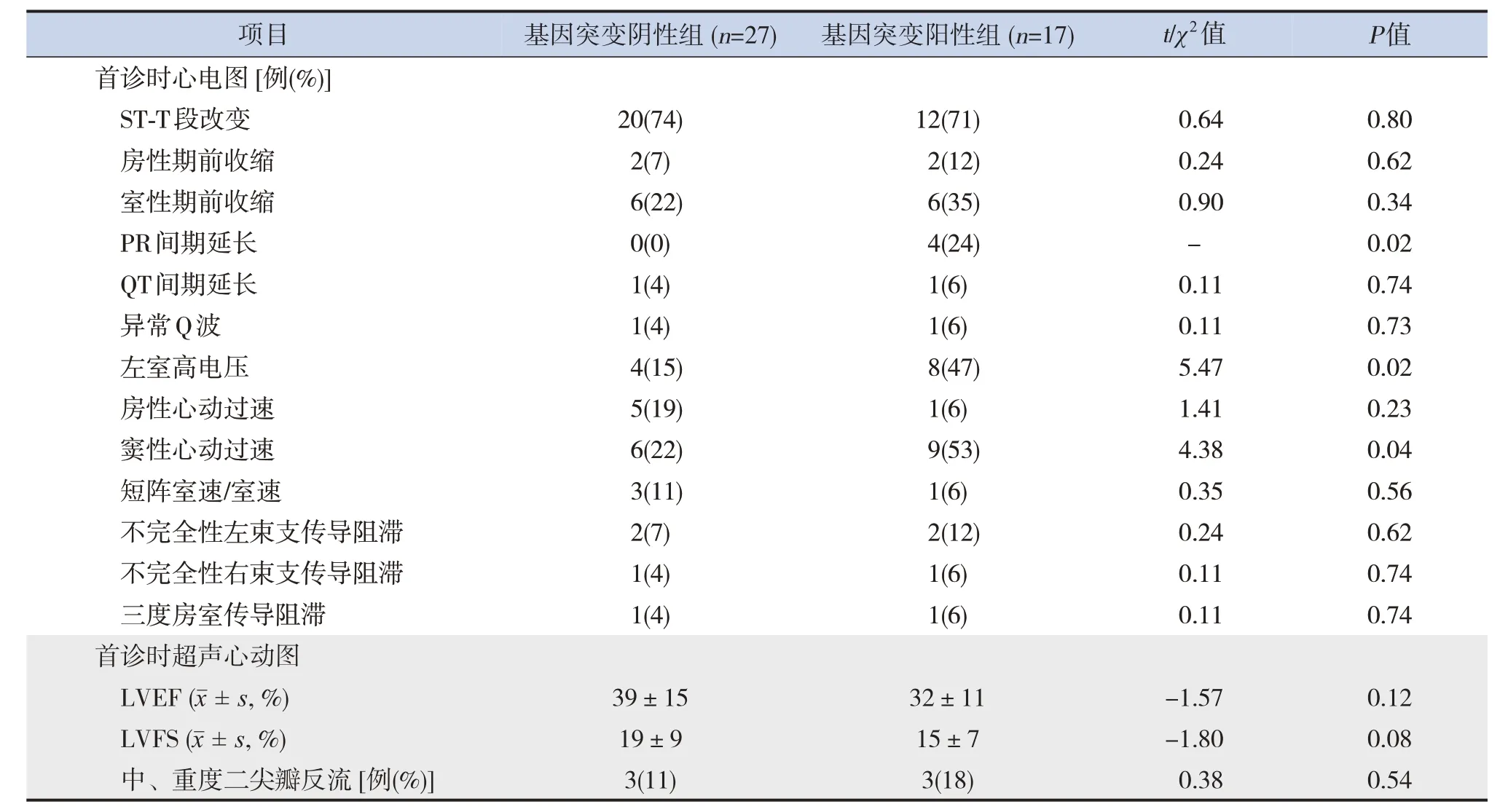
表2 两组首诊时心电图、超声心动图检查结果比较
2.3 基因检测结果
基因突变检出率为39%(17/44),新生突变占24%(4/17)。17例基因突变阳性患儿为TTN基因3例(18%),LMNA、TAZ、MYH7基因各2 例(12%),DMD、PCCB、CTNNA3、TNNI3、FLNC、FBN1、ATAD3A、SGCD基因各1 例(6%)。17 例基因突变阳性DCM患儿首诊时基本资料见表3。中位随访时间为23(8,35)个月,死亡9例(20%)。基因突变阳性组患儿病死率高于基因突变阴性组[47%(8/17)vs 4%(1/27),χ2=12.05,P=0.001]。基因突变阳性组8 例死亡患儿包括3 例TTN、2 例LMNA、2例TAZ和1例ATAD3A基因突变患儿。
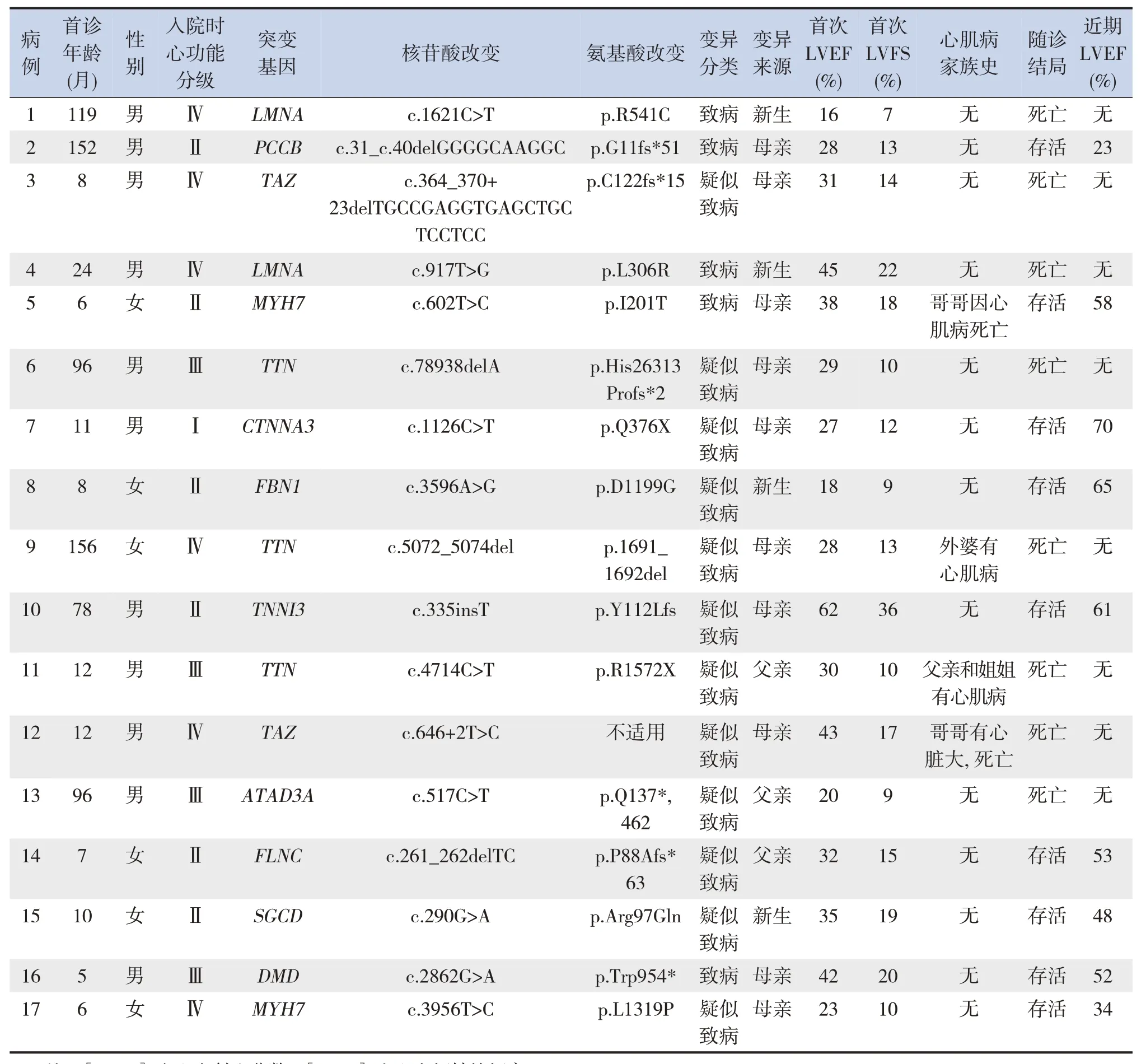
表3 17例基因突变阳性DCM患儿的临床资料
3 讨论
本研究中儿童DCM 基因突变检出率为39%(17/44),与Wang 等[3]报道的约35% DCM 患儿存在致病基因突变相近。基因突变阳性组和阴性组患儿首诊时临床表现、心功能分级Ⅲ~Ⅳ级比例、恶性心律失常、LVEF 和LVFS 等比较差异均无统计学意义,提示DCM 患儿首诊时的严重程度与是否存在致病基因突变无关。本研究中17 例基因突变阳性DCM患儿以TTN基因突变最常见,占18%;其次为MYH7、LMNA和TAZ基因,各占12%。基因突变阳性组患儿病死率高于基因突变阴性组,差异有统计学意义,提示基因突变导致的DCM 患儿预后相对更差。TTN、LMNA、TAZ和ATAD3A基因突变的患儿均在随访期间死亡。
TTN基因突变是DCM 遗传学病因中常见的致病变异,多为TTN截断突变(TTN-truncating variant,TTNtv)。有研究报道在成人DCM 患者中TTNtv占18%~25%,2%~3% 的健康人群携带TTNtv[5]。本研究中TTN基因突变占所有基因突变的18%,在DCM 患儿中的发生率为7%(3/44),与Khan 等[8]研究报道的TTN基因突变发生率为9%相近。TTN基因突变可导致多种骨骼肌病和心肌病,心肌病中以DCM 最常见,其次为肥厚型心肌病(hypertrophic cardiomyopathy,HCM);骨骼肌病包括儿童中央核肌病、Salih 肌病等[5]。同时TTN基因突变可使部分患儿临床表型重叠,而且女性TTNtv携带者有患围生期心肌病的风险[5]。本研究中的3例TTN基因突变患儿均死亡,且3例患儿首诊时LVEF 较低(均≤30%)。Khan 等[8]报道的10 例TTN基因突变DCM 患儿,其中6 例也在首次住院或随诊期间死亡。提示TTN基因突变导致的儿童DCM 往往预后不良,且容易发生恶性心律失常或SCD[3,5]。此外,发病年龄越早或男性的儿童预后相对更差[5]。
MYH7基因突变是HCM 常见的遗传学病因,占30%~50%[9]。1%~5.3%的MYH7基因突变与DCM 表型相关[9]。但本研究中MYH7基因突变占所有基因突变的12%,在DCM 患儿中的发生率为5%(2/44)。MYH7基因突变的不同位置、不同突变类型及遗传模式等均与HCM 患儿的严重程度及临床表型密切相关[9-11]。发生于球状头部及颈部的MYH7基因突变多见于HCM,远端尾部区的突变多与骨骼肌病及DCM表型相关[9-11]。据报道MYH7基因突变相关DCM 表型具有高外显率,多在儿童早期即出现相关表型。van der Meulen等[12]报道过8 例MYH7基因突变DCM 患儿中有6 例为婴儿(<1岁)。本研究中2例MYH7基因突变患儿发病年龄也较早(均<1 岁)。de Frutos 等[10]对106 例MYH7基因突变DCM 患者分析发现,男性发病明显早于女性[(33.3±18.0) 岁vs (41.7±18.6)岁],随访5年约12%的患者预后不良。本研究中2例患儿随访期间均存活,但2例均为女性患儿,其中1 例c.3956T>C(p.L1319P)突变患儿LVEF 值恢复相对较差(近期LVEF 34%);1 例c.602T>C(p.I201T)突变患儿对常规治疗有较好的反应(近期LVEF 58%),但其哥哥因心肌病死亡。提示性别差异可能会影响MYH7基因突变DCM 患儿的预后[9]。
LMNA基因负责编码核纤层蛋白A/C,核纤层蛋白A/C是细胞核膜的主要组成成分。LMNA基因突变可导致DCM,有研究报道LMNA基因突变是DCM 遗传学病因中第2 常见的突变类型,仅次于TTN基因突变,占5%~10%[13]。本研究中LMNA基因突变占所有基因突变的12%,与Ferradini 等[14]报道结果相近。多项研究显示LMNA基因突变是DCM患儿预后不良的标志[1,3,14]。LMNA基因突变相关DCM 是一种高致病性和年龄依赖性的恶性疾病,心脏不良事件发生率高(4 岁时病死率为12%,12 岁时病死率高达30%),同时也是目前指南中唯一获得植入式心律复转除颤器一级预防适应证的基因[15]。LMNA基因突变患者具有较高的房室传导阻滞、室性心动过速、SCD等发生率,且病情进展迅速[16]。本研究中2例LMNA基因突变患儿分别于2岁与9岁发病,且均在首次住院或随访期间死亡。一项包含8 000例成人DCM患者基因检测结果的Meta 分析也显示LMNA基因突变的患者发生心脏不良结局(包括心脏移植、死亡等)的风险显著高于其他突变基因[16]。提示LMNA基因突变相关DCM 患儿多预后不良,其恶性程度相对更高。
TAZ基因突变较为罕见,据报道其突变频率为1/300 000~400 000[17]。TAZ基因突变在本研究中占所有基因突变的12%。TAZ基因突变常导致一种X-连锁隐性遗传的线粒体肌病(Barth 综合征),Barth 综合征患儿多在1 岁内发病,同时约70%的患儿可表现为DCM[18]。本研究中2 例男性患儿均在1岁内发展为DCM,且均在随诊期间死亡。但有部分女性患儿因X染色体倾斜失活而表现出早期发病且严重的DCM表型[17-18]。
DMD基因突变常导致X-连锁隐性遗传的神经肌肉疾病,患儿多在12岁左右发展为DCM[19]。据报道男性新生儿中DMD发病率约为1/3 500[20],但在本研究中较为少见,占所有基因突变的6%。可能与DMD 患儿多在病程早期明确诊断后未能及时到专科门诊随诊或治疗有关。大样本队列研究报道,超过50%的DMD患儿在确诊之后未进行DCM相关治疗或预防治疗[21]。DMD 患儿失访可能低估了DMD基因突变在儿童DCM 遗传学病因中的占比。本研究中还分别发现了ATAD3A、PCCB、FLNC、CTNNA3、FBN1、TNNI3、SGCD基因突变导致儿童DCM各1例。其中1例ATAD3A基因突变的DCM患儿在随诊期间死亡。
综上所述,儿童DCM具有显著的遗传异质性,同一基因突变可导致多种临床表型。本研究显示DCM 患儿首诊时的严重程度与是否存在致病基因突变无关,但致病基因突变导致的DCM 患儿预后相对更差,特别是与TTN、LMNA、ATAD3A和TAZ基因突变相关患儿均在随访期间死亡。不足之处是本研究为单中心、回顾性、小样本研究;个体化治疗可能会影响患儿的预后;随访时间相对较短,远期预后有待进一步追踪;先证者家庭的其他成员未进行基因级联筛查,因此本研究中家族其他成员携带的基因突变可能被低估。今后可开展多中心、大样本研究,以对本研究结果进行验证。
利益冲突声明:所有作者均声明不存在利益冲突。
