黏多糖贮积症的诊治水平现状
2023-07-06邹朝春李正兰戴阳丽潮昀琦
邹朝春 李正兰 戴阳丽 潮昀琦
黏多糖贮积症(mucopolysaccharidosis,MPS)是一组编码酸性黏多糖(又称糖胺聚糖,glycosaminogly‐cans,GAGs)分解代谢的溶酶体酶的遗传缺陷引起的遗传代谢病[1-2]。国外报道MPS 总体发病率约为1/(22 000~25 000)活产儿[1,3],国内尚缺乏相关数据。目前已报道MPS 有8 个类型,12 个相关致病基因,其中MPS ⅣA 和MPS Ⅰ在美国和欧洲国家最常见,国内数据显示MPS ⅣA 和MPS Ⅱ最常见。除了MPS Ⅱ为X 连锁隐性遗传病,其余7 型均为常染色体隐性遗传病[1-2]。目前已知有5 种GAGs,分别为硫酸皮肤素、硫酸乙酰肝素、硫酸软骨素、透明质酸和硫酸角质素。MPS 患儿由于有1 种或多种GAGs 在细胞内积累,干扰细胞正常功能,早期尚处于代偿阶段,出生时均无表型,但随着疾病进展,更多黏多糖累积多系统多脏器,逐步出现无法恢复损伤,严重影响患儿的生活质量和寿命。然而,由于临床对该类疾病认识不足、检测手段缺乏、治疗药物和方法可及性不足,严重制约了对该类疾病的早期筛查、早期诊断和早期治疗。提高MPS 的诊治水平,尤其早期诊治水平,对改善MPS 患儿的预后非常重要。本文就MPS 临床表型、基因型及目前诊治水平作一述评。
1 临床特征
MPS 是一种进行性加重的疾病,患儿出生时通常是正常的,只有当表型随着时间的推移而出现时,才会怀疑患有该疾病。症状出现时患儿年龄越小,往往病情越严重;但出生时具有MPS 样表型的婴儿有可能患有Ⅱ型黏脂贮积症、GM 1 神经节苷脂贮积症或岩藻糖苷贮积症。主要表型如下。
1.1 头面部 面部粗糙、头围大、额头突出、眉毛浓密、多毛症、鼻梁低平、鼻翼肥大、嘴唇厚、舌大、颈短。
1.2 神经系统 出生时神经系统通常是正常的,然后逐渐出现智力、语言、运动和行为的进行性退化。还可能出现攻击性和破坏性行为、自闭症谱系障碍、睡眠障碍、癫痫和其他神经异常(如交通性脑积水、脊髓压迫症状、腕管综合征)。此外,常有齿状突发育不良,并有继发于寰枢椎半脱位的突然且严重脊髓损伤的风险。
1.3 消化系统 腹胀、肝脾肿大和复发性脐疝和(或)腹股沟疝。有些患儿可能会有腹泻或便秘。
1.4 呼吸系统 反复呼吸道感染可能是早期症状之一。上呼吸道阻塞也是早期临床表现,常继发于面中部发育不全、舌大和呼吸道GAGs 积聚。阻塞性睡眠呼吸暂停很常见,一些患儿需要通过鼻罩进行持续气道正压通气。
1.5 心血管系统 可能出现瓣膜性心脏病(通常是二尖瓣和主动脉瓣)、心肌病、心动过速、心律失常、高血压、充血性心力衰竭和外周血管疾病。这是导致死亡的重要原因。
1.6 骨骼和关节 临床表现为关节畸形和脊柱及四肢运动功能障碍。短躯干、短颈、耸肩、脊柱后凸、脊柱侧凸和(或)胸骨突出、骨盆小、髋关节脱位或半脱位、膝外翻、双手腕关节松弛(仅Ⅳ型)、关节挛缩、爪形手、短而粗的手十分常见。患儿通常患有继发于脊柱结构畸形或发育不良的脊髓病。虽然在生命的最初几年生长是正常的,但骨骼发育不良的影响最终会导致生长受限。
1.7 视觉系统 在某些类型中,由于角膜GAGs 沉积出现角膜混浊。也可出现视神经乳头水肿、视神经萎缩和视网膜病变。视觉诱发电位显示视网膜功能下降。其他可有开角型青光眼、白内障和屈光不正。患儿可由于视神经受到压迫出现严重的视力丧失和突然失明。
1.8 耳鼻喉 部分患儿在早期有听力损失(包括感音神经性听力损失和传导性听力损失),而另一些患儿有耳鸣、眩晕、中耳炎、张口受限(由颞下颌关节僵硬引起)、声音粗哑(由喉部GAGs 积聚引起)。
1.9 皮肤 出生后背部和臀部有多个大的“蒙古斑”,这可能是MPS 的早期提示指标。由于GAGs 储存在皮下结缔组织中,面部皮肤明显坚硬厚实,部分患儿出现皮肤结节状或鹅卵石样变化,尤其是肩胛骨、上臂和大腿两侧。
1.10 其他 宽齿距,薄牙釉质,小犬形齿,铲形门牙,龋齿,牙龈增厚也可能发生。
由于MPS 不同类型和亚型的缺陷基因不同,体内蓄积GAGs 类型不同,患儿临床表现也有一定差异,见表1。重度MPS Ⅰ患儿1 岁前反复出现呼吸道感染、脐疝、脊柱后凸畸形。1 岁后面部粗糙和骨骼畸形变得明显。线性生长在3 岁左右减速。神经系统退化(如精神异常)迅速而严重(图1)。轻度MPS Ⅰ可能有轻度神经系统异常,也可能没有神经系统异常。MPS Ⅱ是X 连锁隐性遗传,其症状与MPS Ⅰ相似。MPS Ⅲ有明显的精神发育迟滞、自闭症谱系障碍、行为问题和睡眠障碍突出。MPS Ⅳ一般不影响智力,但骨骼畸形最明显(图2)。MPS Ⅵ一般也不会影响智力。然而,交通性脑积水可能会损害神经系统。MPS Ⅶ可能导致非免疫性胎儿水肿;MPS Ⅸ极其罕见;最近报道了1 个新型,即MPS Ⅹ[4]。

表1 MPS 不同类型和亚型的特点[4-5]

图1 患儿,女,4 岁,MPS Ⅰ型(A:10 月龄时几乎正常;B:1 岁6月龄时出现轻度面部特征和膝外翻;C:2 岁4 月龄时面部特征出现且身高增长停滞)
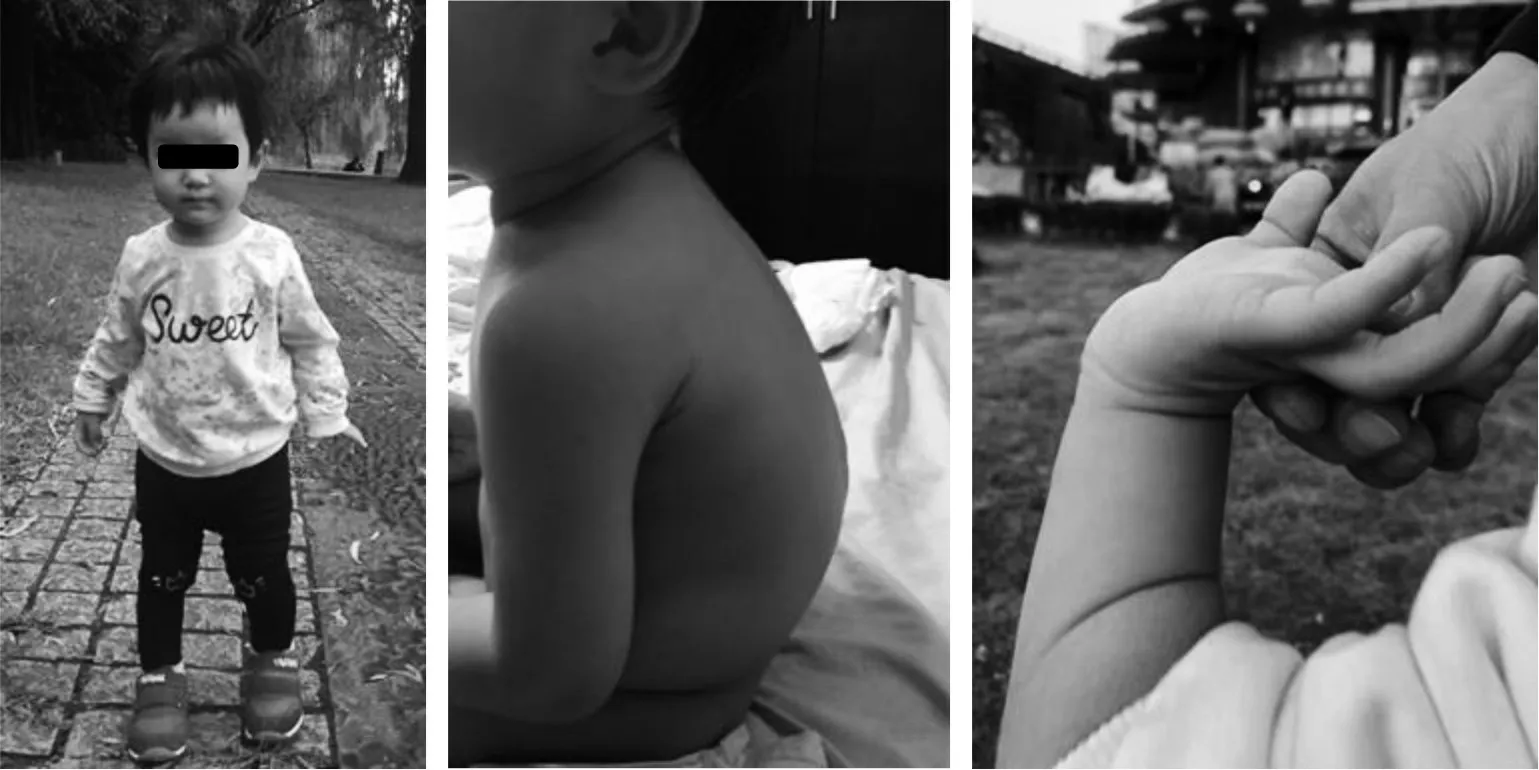
图2 患儿,女,7 岁,MPS ⅣA 型(A:2 岁时面容几乎正常;B:4岁时背部后凸;C:腕关节松弛)
2 诊断和鉴别诊断
对于临床疑似MPS 患儿,可以先进行尿GAGs 检测,确诊主要依靠酶活性和遗传学分析,其中特异性酶学检测被认为是金标准。
2.1 尿GAGs 检测 尿GAGs 检测(如1,9-二甲基亚甲蓝比色检测法)对于筛选MPS 是有用的,尽管定性和半定量测试准确性不足。即使筛查试验结果为阴性,临床医生对于高度疑似患儿也应进一步明确诊断。虽然已经开发了旨在帮助诊断疾病的算法,但由于这类疾病的极端异质性,它们在临床实践中的应用受到限制。串联质谱法(MS/MS)可以检测5 种GAGs 和相关二糖等,并可以区分不同类型的MPS。尿GAGs 也是酶替代疗法(enzyme replacement therapy,ERT)和(或)造血干细胞移植(hematopoietic stem cell transplan‐tation,HSCT)疗效监测的一种方便、廉价的指标。
2.2 酶活性测定 应进行白细胞和血浆中溶酶体酶研究,以明确MPS 的诊断,也有助于明确或排除其他疾病(如寡糖病或半乳糖唾液酸病)。然而,目前国内大多数医院无法对酶活性和尿GAGs 进行检测。
2.3 遗传学分析 目前MPS 的致病基因明确,大部分是点突变。因此,二代测序技术(如全外显子测序)被用作一线遗传学分析方法。值得注意的是,一些基因具有富含GC 的区域和假性基因缺陷。
对于无法通过酶的检测和基因分析确诊的患儿,可考虑皮肤活检检测是否有GAGs 积聚异常。
2.4 产前诊断和新生儿筛查 尽管据报道GAGs 从胎儿阶段就开始沉积,但目前还不建议常规进行产前诊断。在先证者家族中,可以对从绒毛取样和羊膜穿刺术中获得的样本进行基因检测。携带者筛查是降低MPS 患儿出生率的一种新的方法。在美国、我国台湾地区已经将某些类型的MPS 纳入新生儿筛查[6]。我国部分地区已逐步开展新生儿基因筛查,为MPS 的早期诊断提供了可能。
2.5 鉴别诊断 不同类型MPS 的鉴别诊断对特异性治疗很重要。患有Ⅱ型黏脂贮积症、GM 1 神经节苷脂贮积症或岩藻糖苷贮积症的患儿出生时常有MPS 样表型,但是他们的尿GAGs 均无异常。因此,如果尿GAGs 检查呈阴性,应考虑其他诊断可能性,可行尿寡糖和唾液酸分析,以排除寡糖和其他糖蛋白贮积[7-8]。此外,酶分析有助于确认或排除这些疾病。如果所有检测结果都是正常的,需要考虑一些有MPS 类似表现的非溶酶体酶紊乱的疾病(如Coffin-Lowry 综合征)。
3 遗传咨询
受影响个体的子女的患病风险取决于不同类型的MPS 和不同的遗传模式。MPS Ⅱ先证者的男性同胞有50%的风险患有MPS Ⅱ,而女性同胞有50%的风险成为携带者。其他类型MPS 先证者的同胞有25%的风险成为MPS 患儿,50%的风险成为携带者,还有25%的可能性成为正常儿童。
4 治疗和管理
尽管最近治疗方法取得了很大的进展,但对大多数神经系统受到疾病影响的患儿而言,仍无确切的治疗方法。然而,即使在受影响最严重的患儿中,对症治疗也可以对其生活质量产生有益的影响。MPS 是累及多系统的疾病,因此,需多学科、多专业的医生共同参与,进行多学科治疗和管理。
4.1 HSCT 最近,脐带血干细胞(而不是骨髓干细胞)移植在国内被广泛使用。现在普遍认为该疗法用于MPS Ⅲ和MPS Ⅳ患儿的益处较少,建议用于年龄较小(<2~2.5 岁)的重度MPS Ⅰ、MPS Ⅱ和MPS Ⅵ患儿[9-10]。HSCT 成功后,酶活性迅速增加,3~6 个月后尿GAGs 水平降至正常范围,器官肿大和心肌病可能会消失,但角膜通常不会完全恢复至透明。大多数在出生后18 个月内接受HSCT 的MPS ⅠHurler 亚型(重型)患儿可以进一步生长发育,最终发育商(development quotient,DQ)与HSCT 时的DQ 相同。面部骨骼重塑可以改变面部粗糙的外观,使长骨生长更好,明显的脊柱畸形可以随着时间的推移而缓解,但椎体异常没有明显改善,骨骼畸形很难矫正。
4.2 ERT 目前MPS Ⅰ、MPS Ⅱ、MPS ⅣA、MPS Ⅵ和MPS Ⅶ多个ERT 产品已用于临床治疗。在接受治疗的患儿中,耐力(如6 min 步行试验)和呼吸功能得到了改善。在ERT 期间和之后,均要密切观察患儿过敏反应,并做好处理过敏反应的准备。ERT 有不能穿过血脑屏障等局限性。此外,这种治疗非常昂贵,限制了药品临床应用的可及性。
ERT 与HSCT 或基因治疗相结合被认为具有更好的治疗效果和更少的不良反应[10]。
4.3 对症疗法 严重MPS 的对症疗法包括治疗脑积水、中耳疾病和定期止痛治疗关节疼痛和僵硬。这些患儿可能需要神经外科干预以预防颈髓疾病,定期耳鼻喉科、心内科等行矫形评估、心脏状况监测。对于MPS Ⅰ和Ⅵ患儿,眼科随访也至关重要。对于MPS Ⅳ患儿,应加强颈髓和不可逆神经系统疾病的预防。在年纪较大的患儿中,呼吸衰竭管理较为困难,因为患儿对鼻持续通气或双水平气道正压通气反应不佳。
4.4 新疗法研究 目前还有许多其他新疗法正在开发中,从临床前研究到临床试验,如小分子基因激活、基于CRISPR 的激活、寡核苷酸疗法或基于腺相关病毒的基因激活或融合蛋白介导的重组酶[11-12]。
5 小结
MPS 的早期诊断仍然是一个挑战,需要医生对这种罕见疾病有更好的了解。事实上,不是所有的医生,包括儿科医生,都知道这种罕见疾病。此外,大多数新生儿筛查未纳入MPS。尽管ERT 治疗了5 种类型MPS,HSCT 治疗了MPS Ⅰ和MPS Ⅱ,但ERT 的可及性较差,在中国更多的父母选择HSCT。充分认识MPS的发病机制有助于寻找治疗的新靶点和新方法。随着分子遗传学技术的发展,对基因治疗的研究也越来越深入,不过这些研究大多数仍处于非常早期的发现和临床前研究阶段。
(本文由浙江省医学会推荐)
