DNA条形码技术在黑龙江地区姬小蜂科物种鉴定中的应用与分析
2023-07-04邵天玉刘思竹谢维欣刘兴龙李志勇王克勤朱朝东刘丹
邵天玉 刘思竹 谢维欣 刘兴龙 李志勇 王克勤 朱朝东 刘丹

摘要 姬小蜂科寄生蜂在农业害虫防治应用中起着非常重要的作用,但因体型小种类多,鉴定较难,导致开发利用的局限性。随着分子生物学的快速发展,通过形态学与分子生物学相结合的方法来研究小蜂总科已经成为发展趋势。利用黑龙江省的6个吸虫塔收集到了238个姬小蜂科样本。在DNA提取之前通过形态学鉴定为14属52种。对其中60个COI和68个28S基因进行了扩增和测序,并将基因登录号提交GenBank;COI基因的登录号为MG836426~MG836501,28S基因的登录号为MH169011~MH169101。经进一步计算COI和28S基因的种内和种间遗传距离发现,COI基因的种间遗传距离差异显著,最小种间遗传距离(6.00%)远大于最大种内遗传距离(3.02%)。但是,28S基因的差异较大,许多属的种内遗传距离超过种间遗传距离,重叠现象严重。用MEGA-7.0软件进一步分析了COI基因的序列相似性和系统发育关系,结果表明:COI基因序列的聚类结果与形态学分类基本一致,而28S基因序列的聚类结果与形态学结果有较大差异。可见COI基因在姬小蜂科的分类鉴定和系统发育分析上比28S基因有很大优势,更适合姬小蜂科DNA条形码分析。
关键词 DNA条形码;COI;28S;分子鉴定;姬小蜂科
中图分类号 S 433 文献标识码 A 文章編号 0517-6611(2023)05-0078-07
doi: 10.3969/j.issn.0517-6611.2023.05.020
开放科学(资源服务)标识码(OSID):
Application and Analysis of DNA Bar Code Technology in Species Identification of Eulophidae in Heilongjiang Province
SHAO Tian-yu1, LIU Si-zhu1, XIE Wei-xin2 et al
(1.Chongqing University of Posts and Telecommunications, Chongqing 400065;2.Institute of Intelligent System and Bioinformatics, College of Intelligent Systems Science and Engineering, Harbin Engineering University, Harbin, Heilongjiang 150001)
Abstract The Eulophidae parasitoids play a key role in the control of agricultural insects, but because of the small size and many species, identification is difficult, which leads to the limitation of development and utilization. With the rapid development of molecular biology, it’s been quite regular to combine molecular approaches in studying the superfamily Chalcidoidea. In this study, 238 specimens of the family Eulophidae were collected from 6 suction traps in Heilongjiang Province, China. Before DNA extraction, 52 species of 14 genera were identified by morphology. 60 COI and 68 28S genes were amplified and sequenced, which were submitted to GenBank, with accession numbers from MG836426 to MG836501 for COI and from MH169011 to MH169101 for 28S. Intraspecific and interspecific genetic distances were calculated for both COI and 28S genes. The interspecific distances of COI gene was significantly different, with the minimum interspecies genetic distance (6.00%) greater than the maximum intraspecific genetic distance (3.02%), which indicates a clear barcoding gap between species. However, as 28S is highly variable, the intraspecific genetic distances of many genera exceed the interspecific, the overlap phenomenon is obvious. Futher, sequence similarity and phylogenetic relationship were analyzed by MEGA 7.0 software, indicating that the clustering results of COI gene sequences are basically consistent with morphological classification, but that of 28S gene sequences are quite different from those of morphological results. COI gene has some advantages over the conservative 28S gene in the taxonomic identification and phylogenetic resolution of the family Eulophidae, which could be more suitable for DNA barcoding wasps.
Key words DNA barcoding;COI;28S;Molecular identification; Eulophidae
寄生性天敌的正确鉴定对生物防治的高效实施至关重要。姬小蜂科昆虫是世界公认的生物防治的主要寄生蜂之一。但是,由于个体小、种类多、形体多样、鉴定困难,导致了开发利用的局限性。在传统分类鉴定上,要想识别来自某个地区或某个项目的姬小蜂科昆虫,需要专业领域的、甚至是从事昆虫分类的专家学者才能实现。但由于传统分类的形态学人工鉴定效率很低,已无法满足当前生物防治快速发展和大规模识别工作的需求。
DNA条形码已被广泛应用于识别全球的生物物种,也逐渐被认为是行之有效的通用分类方法[1-6]。
该研究将收集到的姬小蜂科样本,通过基于线粒体COI 和28S基因的DNA条形码技术,与NCBI(https://www.ncbi.nlm.nih.gov/genbank/)系统进行比对鉴定,测试物种鉴定的成功率。此外,通过计算COI和28S基因种内和种间遗传距离,检验了DNA条形码技术在姬小蜂科昆虫鉴定中的准确性。建立了黑龙江地区姬小蜂科DNA条形码数据库,补充了姬小蜂科寄生性天敌物理种的地理资源信息。
1 材料与方法
1.1 样本取样
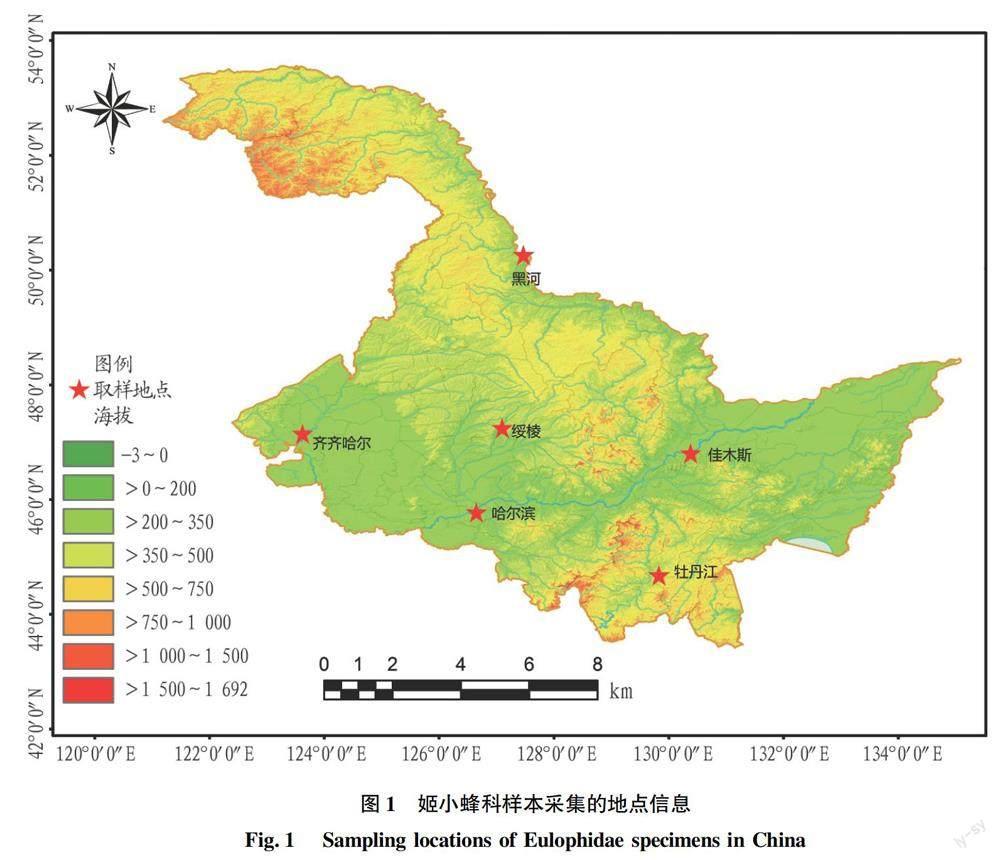
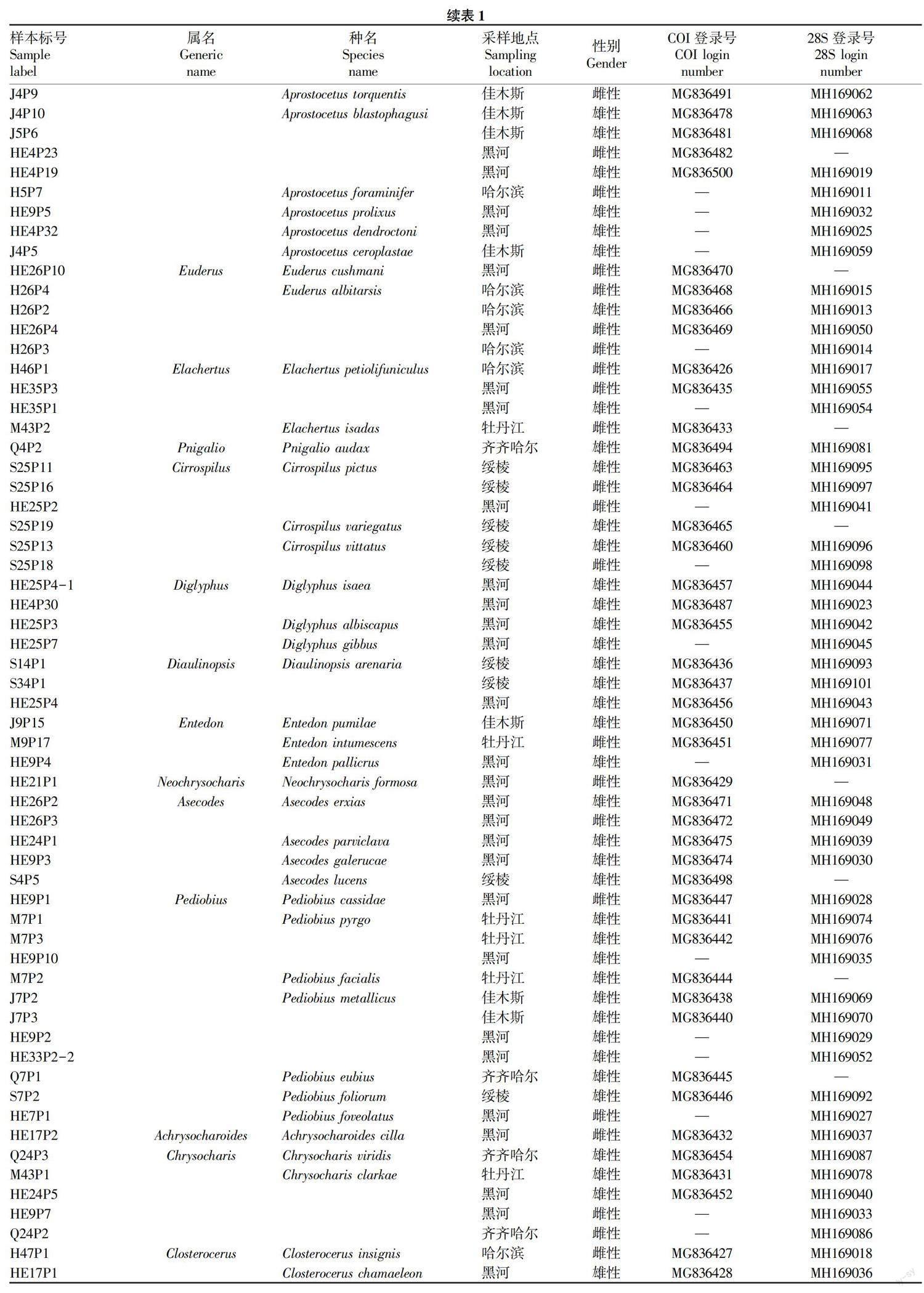
利用黑龙江省的6个吸虫塔收集了238个姬小蜂科样本,均保存于无水乙醇中。采样位置、种类名称和GenBank登录号的详细信息见图1和表1。在DNA提取之前,依据形态学鉴定为14属52种[7-20]。所有标本存放在黑龙江省农业科学院植物保护研究所。
1.2 DNA提取、扩增和测序
使用QIAGEN DNeasy提取试剂盒提取基因组DNA。COI基因使用LA Taq(TAKARA)扩增, 28S使用MightyAmp(TAKARA)扩增。COI基因使用条形码引物LCO1490(5′-GGTCA ACAATCAATCAAATTGG-3′) 和HCO2198(5′-TAAACTTCAGGTGACCAAAAATCA-3′)进行扩增。28S基因使用条形码引物D2-3549F(5′-AGTCGTGTTGTGTGTGCAG-3′)和D2-4068R(5′-TTGGTCGTTTCAAGC-GGG-3′)进行扩增。所有扩增反应均采用50 μL的PCR体系。COI反应体系:5.0 μL 10×LA缓冲液、5.0 μL MgCl2(2.5 mmol/L)、5.0 μL dNTP(2.5 mmol/L)、1.0 μL正反向引物(10 mmol/L)、0.5 μL LA Taq聚合酶(5 U/μL)、2.0~4.0 μL模板DNA和蒸馏水,最终PCR体系补充为50 μL。28S反应体系:25.0 μL Mightymp缓冲液、2.1 μL mightyMP DNA聚合酶(1.25 U/μL),正反向引物1.0 μL(10 mmol/L),2.0~4.0 μL模板DNA和蒸馏水,最终PCR体系补充为50 μL。COI PCR反应条件为94 ℃预变性2 min,35个循环包括:94 ℃30 s,48~50 ℃50 s,72 ℃延伸1 min,72 ℃延伸10 min。28S PCR反应条件为98 ℃预变性2 min,35个循环包括:98 ℃10 s,58 ℃15 s,68 ℃延伸1 min,68 ℃延伸5 min。用ABI3130测序仪进行测序。
1.3 序列比對与分子分析
将测序结果导入DNAStar中的SeqMan软件[21],进行序列的拼接与手工校正,确定分析的序列,利用NCBI中的“BLAST”软件进行相似性检索,确定序列方向及目的片段;将确定的序列以及GenBank下载相应序列载入Clustal X 1.83 软件[22] 进行序列比对,输出格式为FASTA。比对结果导入MEGA 7.0 软件[23],计算各物种间的遗传距离,转换和颠换值及其比值(R值),保守位点(conserved sites,C)及变异位点(variable sites,V)等数值。
采用MEGA 7.0软件统计序列碱基组成、GC含量、多态位点和简约信息位点等参数;利用Kimura 2-parameter(K2-P)双参数模型计算群体内和群体间遗传距离;以广赤眼蜂Trichogramma evanescens Westwood(Trichogrammatidae)作为外群,采用邻接法(neighbor-joining,NJ)构建系统发育进化树。
2 结果与分析
2.1 NCBI系统的识别结果
该研究获得了60个625 bp的COI序列和68个620 bp的28S序列。所有DNA序列均已提交给GenBank,登录号分别为MG836426~MG836501和MH169011~MH169101。
根据COI基因序列,可以直接鉴定出4属5种,分别为S25P13、HE25P4-1、HE4P30、HE21P1和HE17P2,相似性分别为95%、98%、98%、98%和94%,物种鉴定率为8.3%,按属分类共鉴定出38个样本,占所有样本的63.3%。此外,共有17个样本被错误识别,占所有样本的28.3%。根据28S基因序列,可以直接鉴定出2属5种,分别为H26P4、H26P2、HE26P4、H26P3和HE17P2,所有相似性均为99%,物种鉴定率为7.35%。根据属分类,共鉴定出51个样本,占所有样本的75.0%。此外,共有12个样本被错误识别,占所有样本的17.6%。
2.2 遗传距离分析
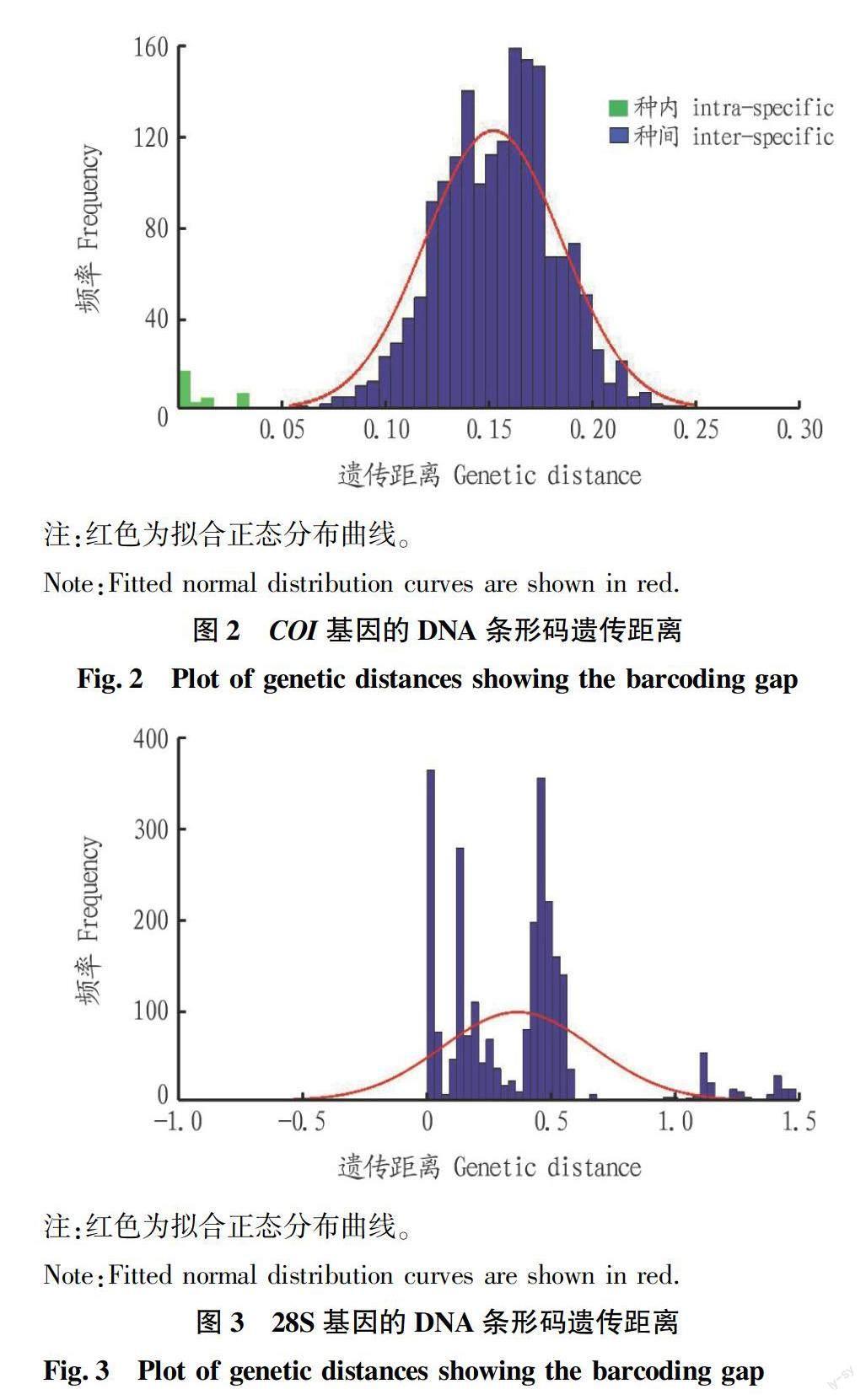
通过对黑龙江省姬小蜂科样本COI和28S基因的遗传距离分析可知,COI基因的种间平均遗传距离(15.40%)是种内平均遗传距离(0.60%)的25.67倍,而28S基因的种间平均遗传距离(64.8%)是种内平均遗传距离(0.27%)的240倍。这完全符合DNA条形码有效性的标准要求:种间平均遗传距离至少是种内平均遗传距离的10倍以上[24]。
基于COI基因平均遗传距离计算可知,种间种内平均遗传距离差异显著。最小种间遗传距离(6.00%)大于最大种内遗传距离(3.02%),不同物种之间没有重叠,DNA条形码显示出较大的种间距离和较小的种内距离,存在明显的条形码缺口(图2)。然而,28S基因是异常多变的。多个属的种内遗传距离远超过种间遗传距离。重叠现象明显,种内和种间物种之间没有明显的“条形码差异”(图3)。
2.3 系统发育分析
经计算可知,COI基因序列中A的平均频率为30.8%,C、G、T的平均频率分别为12.7%、13.8%、42.7%。COI基因序列显示出强烈的腺嘌呤和胸腺嘧啶(AT)偏好,高达73.5%,这是昆虫线粒体基因组的典型特征[25-26]。而28S基因序列中A、C、G、T的平均频率分别为18.5%、27.1%、32.4%、22.0%。
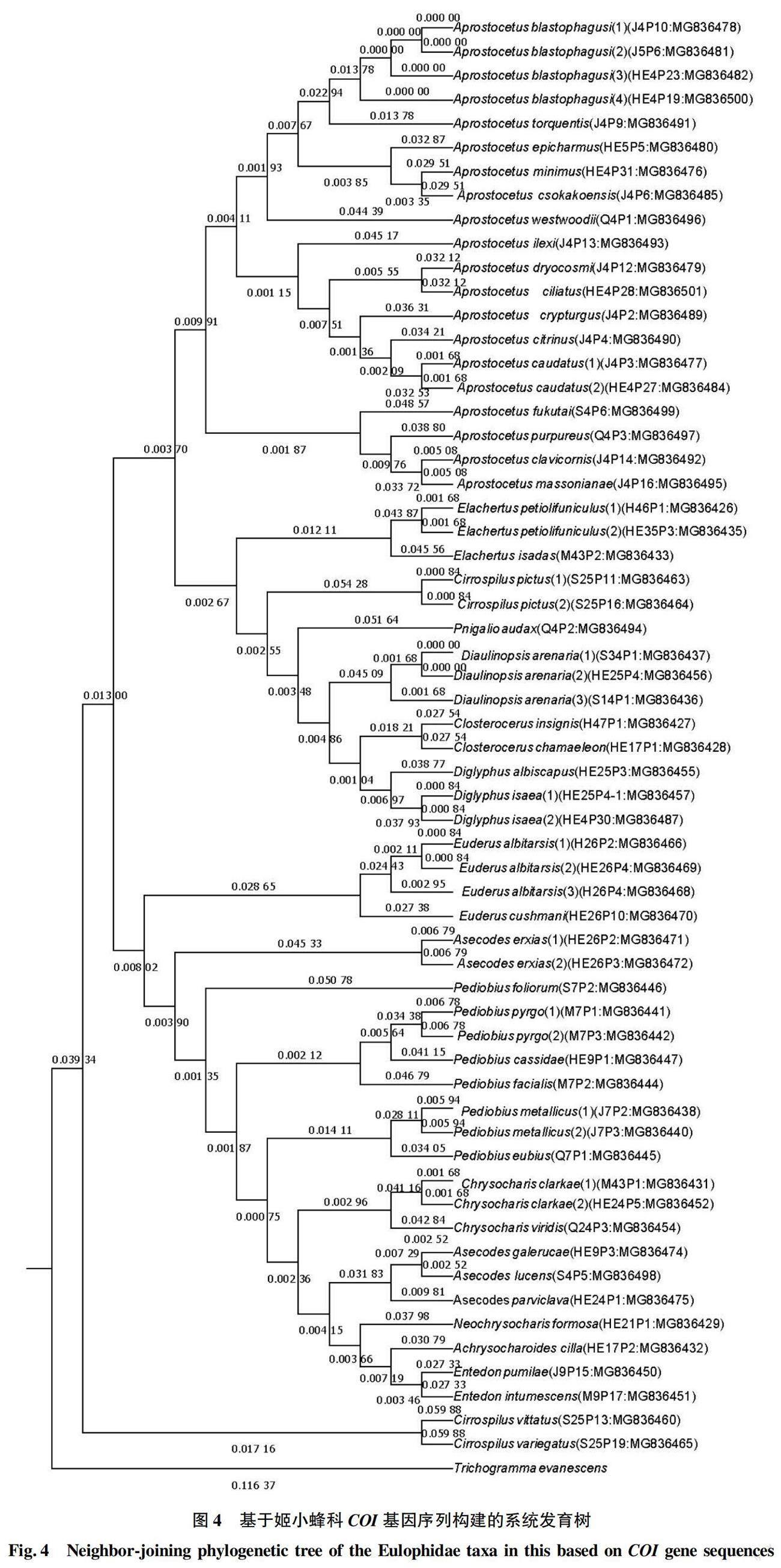
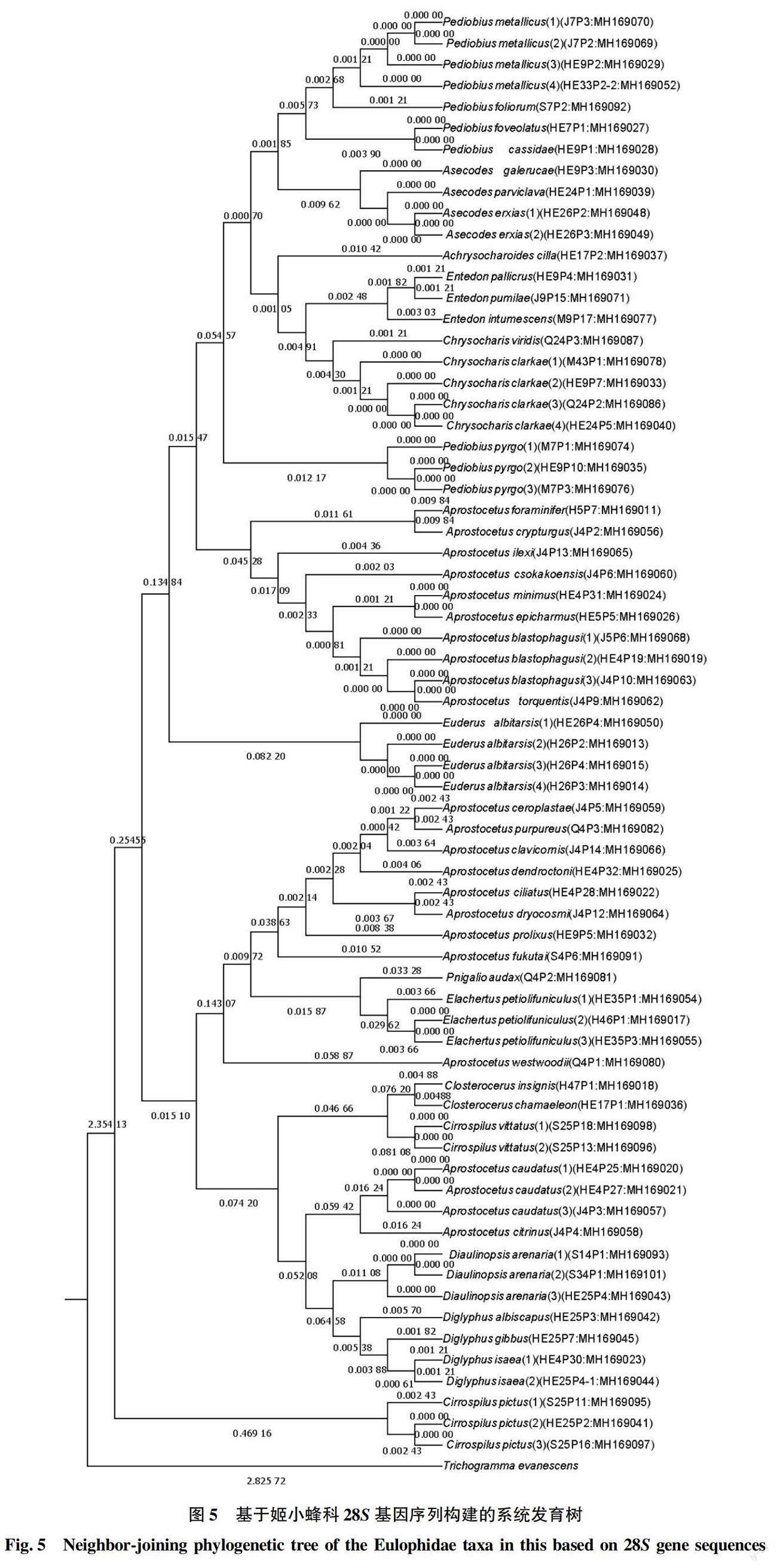
基于COI基因序列的MP系统发育树可知,53个样本同聚为一个单系,占88.3%,而该科的所有样本又同聚集成一个系,将广赤眼蜂作为外群,聚类结果基本符合形态学分类(图4)。然而28S基因序列的聚类结果与形态学分类结果有较大差异,只有29个样本同聚集成一个单系,占42.7%。28S基因序列的种内遗传距离甚至比种间遗传距离还大(图5)。
3 结论与讨论
在该研究中,DNA条形码在姬小蜂科鉴定中的作用仍然有限。部分样本只能进行属级鉴定,物种鉴定必须结合形态学特征。但通过DNA条形码的初步鉴定,确实可以有效地缩小鉴定范围,大幅减少全部基于形态学特征的鉴定和验证所需的精力和时间,进一步确保所有样品的准确识别。
DNA条形码技术广泛应用于生态学、生物多样性、检验检疫和法医鉴定等[27-28],为诸多研究领域都提供了便利。DNA条形码技术在生物分类鉴定中发挥着越来越重要的作用,是一种快速有效的鉴定工具,逐步得到各研究领域更多学者的支持[29]。在昆虫分类学研究中,DNA条形码不受样本条件的限制,对受试者的形态完整性更没有严格要求,可以大量且同时进行,大大简化了物种鉴定过程[30-31]。然而, DNA条形码不能完全取代形态学分类,对于部分昆虫分类,它仅可以作为形态学鉴定的辅助工具,为传统形态学分类提供分子基础[32]。因此,笔者将形态分类学和分子分类学相结合,对黑龙江省姬小蜂科的种类进行研究,大大提高了种类鉴定结果科学性和准确性。
研究报道,COI基因序列的平均种间遗传距离小于28S基因序列是由于28S基因的進化速度相对较慢,种间高度保守,更适合属间的系统发育分析[33]。COI基因系单亲遗传,没有重组,而且进化速度相对较快[1],种内遗传变异比28S基因相对较低,物种特异性较高,更适合分析物种、亚种和近缘物种[34]。与该研究中,COI基因序列比28S基因序列构建的系统发育树更接近于传统的形态分类,COI基因序列更适合用于姬小蜂科的分子鉴定、系统发育和DNA条形码标记的结论相互印证。
随着分子测序技术的发展,高通量测序技术已被应用于获取DNA条形码序列,从而有可能识别大量物种[35-36]。希望增加基因数量可以构建更精确的系统发育树[37],而多个基因的组合可以在系统分析中提供更有效的系统发育信息[38-39]。基于多基因片段组合的DNA条形码识别系统将成为一种探索性的趋势,未来的DNA条形码分析研究将朝着多基因、多方法、多学科组合的方向发展,只有通过有效的序列分析才能获得科学准确的结果。
参考文献
[1] HEBERT P D,RATNASINGHAM S,DEWAARD J R.Barcoding animal life:Cytochrome c oxidase subunit 1 divergences among closely related species[J].Proceedings of the royal society B:Biological sciences,2003,270(S1):S96-S99.
[2] EBACH M C,HOLDREGE C.DNA barcoding is no substitute for taxonomy[J].Nature,2005,434(7034):697.
[3] SCHINDEL D E,MILLER S E.DNA barcoding a useful tool for taxonomists[J].Nature,2005,435(7038):17.
[4] DINC V,ZAKHAROV E V,HEBERT P D N,et al.Complete DNA barcode reference library for a country’s butterfly fauna reveals high performance for temperate Europe[J].Proceedings of the royal society B:Biological sciences,2011,278(1704):347-355.
[5] WANG Y,ZHOU Q S,QIAO H J,et al.Formal nomenclature and description of cryptic species of the Encyrtus sasakii complex(Hymenoptera:Encyrtidae)[J].Scientific reports,2016,6:1-16.
[6] 周诗语.中国迈金小蜂属分子系统学研究[D].沈阳:沈阳师范大学,2018:1-84.
[7] YOSHIMOTO C M.Revision of the genus Chrysocharis Frster(subgenus Chrysocharis s.str)(Eulophidae:Chalcidoidea)of America north of Mexico[J].The Canadian entomologist,1973,105(11):1377-1405.
[8] ZHU C D,LASALLE J,HUANG D W.A study on Chinese species of Aulogymnus Frster(Hymenoptera:Eulophidae)[J].Insect sinica,1999,6(4):299-308.
[9] ZHU C D,LASALLE J,HUANG D W.A review of the Chinese Diglyphus Walker(Hymenoptera:Eulophidae)[J].Oriental insects,2000,34(1):263-288.
[10] ZHU C D,HUANG D W.A taxonomical study of Eulophidae in Zhejiang,China((Hymenoptera:Chalcidoidea)[J].Acta zootaxonomica sinica,2001,26(4):533-547.
[11] ZHU C D,HUANG D W.A study of Chinese Elachertus Spinola(Hymenoptera:Eulophidae)[J].Zoological studies,2001,40(4):317-354.
[12] ZHU C D,LASALLE J,HUANG D W.A study of Chinese Cirrospilus Westwood(Hymenoptera:Eulophidae)[J].Zoological studies,2002,41(1):23-46.
[13] ZHU C D,HUANG D W.A taxonomical study on Eulophidae from Guangxi,China(Hymenoptera:Chalcidoidea)[J].Acta zootaxonomica sinica,2002,27(3):583-607.
[14] ZHU C D,HUANG D W.Review of Chinese species of genus Eulophus(Hymenoptera:Eulophidae)[J].Entomological news,2002,113(1):50-62.
[15] HUANG D W,ZHU C D.Revision of Chinese Euplectromorpha Girault((Hymenoptera:Eulophidae)[J].Insect systematics and evolution,2000,31(4):401-410.
[16] ZHU C D,HUANG D W.Preliminary cladistics and review of Hemiptarsenus Westwood and Sympiesis Frster(Hymenoptera:Eulophidae)in Hungary[J].Zoological studies,2000,42(2):307-335.
[17] ZHU C D,HUANG D W.A study of the genus Euplectrus Westwood(Hymenoptera:Eulophidae)in China[J].Zoological studies,2003,42(1):140-164.
[18] ZHU C D,HUANG D W.Chinese species of Diglyphomorphomyia Girault(Hymenoptera:Eulophidae)[J].Zoological studies,2003,42(3):444-449.
[19] PROTASOV A,BLUMBERG D,BRAND D,et al.Biological control of the eucalyptus gall wasp Ophelimus maskelli(Ashmead):Taxonomy and biology of the parasitoid species Closterocerus chamaeleon(Girault),with information on its establishment in Israel[J].Biological control,2007,42(2):196-206.
[20] CAO H X,LA SALLE J,ZHU C D.Chinese species of Pediobius Walker (Hymenoptera:Eulophidae)[J].Zootaxa,2017,4240(1):1-71.
[21] PARCHMAN T L,GEIST K S,GRAHNEN J A,et al.Transcriptome sequencing in an ecologically important tree species:Assembly,annotation,and marker discovery [J].BMC genomics,2010,11:1-16.
[22] CHENNA R,SUGAWARA H,KOIKE T,et al.Multiple sequence alignment with the Clustal series of programs[J].Nucleic acids research,2003,31(13):3497-3500.
[23] KUMAR S,NEI M,DUDLEY J,et al.MEGA:A biologist-centric software for evolutionary analysis of DNA and protein sequences[J].Brief bioinform,2008,9(4):299-306.
[24] HEBERT P D N,CYWINSKA A,BALL S L,et al.Biological identifications through DNA barcodes[J].Proceedings of the royal society of London B:Biological sciences,2003,270(1512):313-321.
[25] SIMON C,FRATI F,BECKENBACH A,et al.Evolution,weighting,and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers[J].Annals of the entomological society of America,1994,87(6):651-701.
[26] ARIAS M C,SHEPPARD W S.Molecular Phylogenetics of honey bee subspecies(Apis mellifera L.)inferred from mitochondrial DNA sequence[J].Molecular phylogenetics and evolution,1996,5(3):557-566.
[27] PEI N C.Identification of plant species based on DNA barcode technology[J].Chinese journal of applied ecology,2012,23(5):1240-1246.
[28] 趙广宇,李虎,杨海林,等.DNA条形码技术在昆虫学中的应用[J].植物保护学报,2014,41(2):129-141.
[29] LI Q Q,LI D Y,DUAN Y Q,et al.Application of DNA barcoding in lepidopteran insects[J].Chinese bulletin of life sciences,2010,22(4):307-312.
[30] SHUFRAN K A,PUTERKA G J.DNA barcoding to identify all life stages of holocyclic cereal aphids(Hemiptera:Aphididae)on wheat and other Poaceae[J].Annals of the entomological society of America,2011,104(1):39-42.
[31] EKREM T,WILLASSEN E,STUR E.A comprehensive DNA sequence library is essential for identification with DNA barcodes[J].Molecular phylogenetics and evolution,2007,43(2):530-542.
[32] JIN Q,CHEN F,LUO G J,et al.Estimation of species richness of moths(Insecta:Lepidoptera)based on DNA barcoding in Suqian,China[J].Biodiversity science,2016,24(11):1296-1305.
[33] HOLTERMAN M,VAN DER WURFF A,VAN DEN ELSEN S,et al.Phylum-wide analysis of SSU rDNA reveals deep phylogenetic relationships among nematodes and accelerated evolution toward crown clades[J].Molecular biology & evolution,2006,23(9):1792-1800.
[34] JIANG L S,LI Q,KONG L F.Phylogenetic relationships among Sanguinolaria species in the coastal waters of China[J].Journal of fishery sciences of China,2018,25(5):936-948.
[35] ZHOU X,LI Y Y,LIU S L,et al.Ultra-deep sequencing enables high-fidelity recovery of biodiversity for bulk arthropod samples without PCR amplification[J].GigaScience,2013,2(1):1-12.
[36] CAO Y,SHEN W J,CHEN L,et al.Application of metabarcoding technology in studies of fungal diversity[J].Biodiversity science,2016,24(8):932-939.
[37] WOLF Y I,ROGOZIN I B,GRISHIN N V,et al.Genome trees constructed using five different approaches suggest new major bacterial clades[J].BMC evolutionary biology,2001,1(1):1-22.
[38] GIRIBET G,EDGECOMBE G D,WHEELER W C.Arthropod phylogeny based on eight molecular loci and morphology[J].Nature,2001,413(6852):157-161.
[39] GRANDJEAN F,TAN M H,GAN H M,et al.Rapid recovery of nuclear and mitochondrial genes by genome skimming from Northern Hemisphere freshwater crayfish[J].Zoologica scripta,2017,46(6):718-728.
