血管性血友病的分子机制及诊治研究进展
2023-06-28罗婧媛综述审校
罗婧媛 综述,陈 姝 审校
重庆医科大学附属第二医院血液内科,重庆 400010
血管性血友病(VWD)是人类最常见的常染色体遗传性出血性疾病,由血管性血友病因子(VWF)基因突变导致VWF量(1型和3型VWD)或质(2型VWD)的缺陷引起,以皮肤黏膜出血和创伤或侵入性手术后的过度出血为主要临床表现,严重者可发生胃肠道或关节肌肉出血。VWD复杂的分子病理机制使其诊断和治疗一直是临床上的难题。近年来的研究增强了对VWD发病机制的理解,同时开发出了新的诊断方法和治疗手段。本文对 VWD 的分子基础、诊断分型和治疗的研究现状及进展综述如下。
1 VWF的生物学特点
1.1VWF的生物合成和分子结构 VWF是由血管内皮细胞和骨髓巨核细胞合成的一种多聚体糖蛋白,对血小板黏附于暴露的内皮下胶原、血小板聚集和凝血因子Ⅷ(FⅧ)的稳定至关重要,在生理性止血和血栓形成过程中起着重要作用[1]。VWF基因位于12号染色体,由52个外显子和51个内含子组成,编码含2 813 个氨基酸残基(aa)的前体蛋白,包括22个aa的信号肽(SP),741个aa的前肽(VWFpp)和2 050个aa的成熟亚单位,结构域组成为D1-D2-D′-D3-A1-A2-A3-D4-C1-C2-C3-C4-C5-C6-CK[2],功能结构域包括血小板糖蛋白Ⅰb(GPⅠb)结合位点(A1结构域)、蛋白裂解位点(A2结构域)、胶原结合位点(A1和A3结构域)、FⅧ结合位点(D′和D3结构域)及血小板糖蛋白ⅡbⅢa(GPⅡb/Ⅲa)结合位点(C4结构域),见图1。
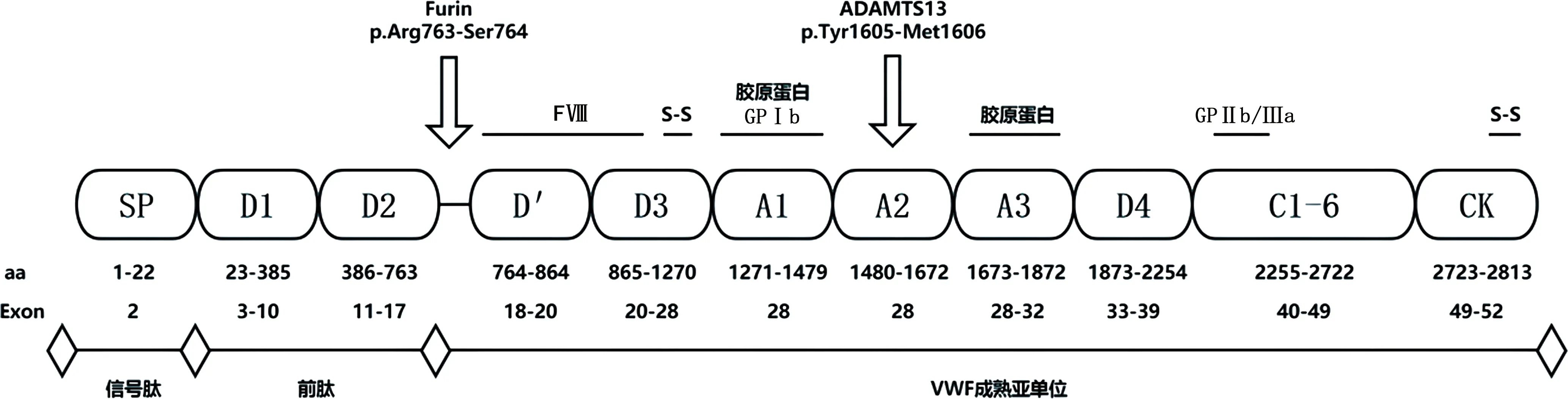
注:Exon为外显子。图1 VWF前体蛋白的结构和功能以及编码基因
VWF前体蛋白在核糖体中翻译完成,去除信号肽后转运至内质网,CK结构域之间通过二硫键(S-S)连接形成VWF二聚体,后者转运至高尔基体,D3结构域之间通过S-S连接形成VWF多聚体。VWFpp和D′结构域使二聚体正确对齐,同时VWFpp通过其CGLC序列的蛋白二硫化物异构酶活性催化D3结构域之间的S-S形成[3],这对于多聚化至关重要。多聚体生成后,VWFpp被furin蛋白酶裂解,但仍与D′D3结构域保持高亲和力的非共价结合[4]。
1.2VWF的储存、分泌和代谢 新生的VWF多聚体先被运输至内皮细胞的Weibel-Palade小体(WPBs)和巨核细胞/血小板的α-颗粒中储存,后续通过基础型、调节型和组成型3种分泌方式释放入血[5-6]。多聚化程度越高的VWF具有越强的促血小板黏附和促血栓形成能力,金属蛋白酶ADAMTSL3通过裂解高分子量的VWF多聚物(VWF-HMWMs)重塑了VWF多聚体的大小分布,从而防止VWF-HMWMs诱导的血小板过度聚集和血栓形成[7]。VWF主要被巨噬细胞内吞清除,半衰期为8~12 h,健康人血浆VWF水平在50~200 IU/dL波动[8]。VWFpp与成熟VWF等比例地分泌入血,但其清除独立于VWF,半衰期为2~3 h,水平约为100 IU/dL[4]。VWFpp与VWF在体内等比例分泌而代谢途径不同,因此VWFpp/VWF抗原(VWF:Ag)可以反映体内VWF的清除情况,健康人VWFpp/VWF:Ag<3.0[9],VWFpp/VWF:Ag增加提示VWF清除增强。
2 VWD 的分型及分子机制
VWD可分为3种类型:1型为VWF量的部分缺失,2型为VWF 质的缺陷伴或不伴量的缺乏,3型为VWF量的完全缺失。
2.11型VWD 1型VWD为常染色体显性遗传,以VWF数量轻度或中度减少,但VWF功能及多聚体分布基本正常为特征,发病率最高,约占所有患者的75%。人类基因突变数据库(HGMD)中记录了超过250种不同的1型VWD基因突变,大多数发生在28号外显子,约80%为错义突变,其余为小缺失/插入、剪切突变、无义突变、启动子突变、基因转换等,所占比例均<10%。分子致病机制包括以下3种:(1) VWF合成减少。低VWF产量一般与杂合性无效等位基因相关。无义突变、剪切突变、缺失和插入等均可产生无效等位基因,不能合成有功能的蛋白质[10],VWF生成仅来自非突变的等位基因,导致VWF产量下降50%,如p.Arg1659X和p.Arg1853X;启动子突变破坏转录因子结合,VWF基因转录衰减,蛋白生成减少。(2)VWF分泌受损。部分突变改变VWF前体蛋白结构,内质网质量控制系统阻止缺陷蛋白向高尔基体转运,VWF滞留在内质网中或在细胞内被降解[10]。所有破坏链内S-S的特异性半胱氨酸突变都可能改变VWF构象而导致这种细胞内滞留[11],如p.Cys1060Tyr、p.Cys1130Phe、p.Cys1149Arg、p.Tyr1584Cys、p.Cys2257Ser和p.Cys2671Tyr。p.Tyr1584Cys是最常见的1型VWD突变,位于A2结构域,可在约15%的VWD患者中检测到杂合子,此类患者平均VWF:Ag为40 IU/dL,血型多为O型。p.Tyr1584Cys不仅导致VWF内质网滞留,还会使VWF对ADAMTS13的敏感性增加[12]。部分突变干扰WPBs形成而使VWF基础型和调节型分泌减少[13-14],如p.Cys2190Tyr和p.Ala1716Pro。(3)VWF清除增强。在>40%的1型VWD患者中观察到病理学增加的VWF清除率,将这种VWF清除增强所导致的1型VWD称为1C型。此类突变主要位于D3结构域,如p.Arg1205His/Cys/Ser,p.Cys1130Phe/Gly/Arg,p.Trp1144Gly和p.Cys1149Arg,这提示D3结构域可能包含VWF存活和清除的调控或识别位点。此外,位于D4结构域的p.Ser2179Phe和位于C6结构域的p.Cys2671Tyr也与VWF 清除增加有关[10]。VWD Vicenza(p.Arg1205His 杂合突变)是1C型VWD的经典类型,VWF:Ag通常为10~15 IU/dL,VWFpp/VWF:Ag显著升高(通常>10)并伴有VWF-HMWMs的轻微增加。研究表明p.Arg1205His通过增强VWF与肝脾巨噬细胞受体的结合提高清除率,因此有专家推荐将VWD Vicenza归类为2型VWD(而不是1型),以强调与清除受体更强的相互作用是一种新的VWF功能缺陷[15]。
2.22型VWD 2型VWD以VWF功能异常为特征,占所有VWD患者的20%~30%,基于VWF与血小板GPⅠb的结合异常进一步分为2A、2B和2M型,与FⅧ的结合缺陷分为2N型。大多数2型VWD为显性遗传,但2N型和部分2A型为隐性遗传。
2.2.12A型VWD VWF依赖的血小板黏附功能主要由VWF-HMWMs介导。2A型VWD的发病机制为VWF-HMWMs分泌减少或裂解增加,选择性缺乏VWF-HMWMs导致的VWF-血小板结合活性下降。突变类型有错义、插入、缺失、移码突变,大多数为错义突变且位于A2结构域,集中在ADAMTS13酶解位点Tyr1605-Met1606周围、Glu1504-Lys1672范围内,导致2种不同的致病机制:(1)改变A2 区构象使ADAMTS13切割位点(Tyr1605-Met1606)易于暴露,VWF对ADAMTS13的敏感性增加,VWF-HMWMs裂解增强[12],如p.Gly1505Glu、p.Met1528Val、p.Arg1597Trp,p.Val1607Asp、p.Gly1609Arg、p.Gly1629Glu、p.Gly1631Asp和p.Glu1638Lys;(2)损害VWF在细胞内的合成和加工,蛋白缺陷而滞留于细胞内,VWF多聚体特别是VWF-HMWMs分泌减少[16],如p.Gly1505Arg、p.Ser1506Leu、p.Leu1540Pro、p.Val1607Asp。值得注意的是,p.Gly1505Glu与p.Gly1505Arg发生在同一个密码子上,这说明A2结构域中氨基酸替换的位置与2A型VWD的分子致病机制没有关联。此外,CK结构域的突变可影响VWF二聚体化,如p.Cys2771Arg和p.Cys2773Arg;D3结构域的突变可影响VWF多聚体化,如p.Cys1099Tyr、p.Cys1143Tyr和p.Cys1173Arg:上述突变均为半胱氨酸残基被替换,导致二聚化或多聚化过程中所必需的分子间S-S生成障碍,VWF-HMWMs合成和分泌减少[17]。A1结构域的突变也可使VWF多聚化受损,但机制尚不明确,同时常伴有VWF与血小板GPⅠb结合亲和力的增强或降低,如p.Cys1272Arg/Gly、p.Val1314Phe、p.Arg1315Cys和p.Cys1458Tyr。A2、CK、D3和A1结构域的突变均为显性突变。D2结构域的突变为隐性突变,破坏VWFpp构象而干扰VWF多聚体形成,如错义突变p.Asn528Ser、 p.Gly550Arg和p.Cys623Trp,插入突变p.Phe404_Thr405insAsnPro和 p.Asn624_Ala625insGly,以及缺失突变p.Cys709LeufsTer711。造成VWF生物合成缺陷、细胞内滞留,多聚体不能正常分泌的2A型突变为GroupⅠ突变;提高VWF-HMWMs对ADAMTS13的敏感性,使其裂解增加的突变为Group Ⅱ突变。GroupⅠ突变会导致比GroupⅡ突变更加严重的出血表现,但Group Ⅰ突变的VWD患者对去氨加压素(DDAVP)治疗的反应更好[12,18]。
2.2.22B型VWD 2B型VWD的发病机制为VWF-HMWMs与血小板GPⅠb的亲和力增强,在体内自发性形成VWF-血小板复合物后被清除,因此2B型患者还常有不同程度的间歇性血小板减少,可因应激而加重,特别是妊娠期间经常发生严重的血小板减少,婴儿往往有新生儿血小板减少症[19]。VWF与血小板GPⅠb的结合位点位于A1结构域。2B型突变为A1内的功能获得性突变,稳定A1的结合构象使VWF与GPⅠb的结合能力增强。2B型突变大部分为错义突变,主要在Cys1272-Cys1458二硫环中,Arg1306、Arg1308、Arg1341为突变热点[20],p.Arg1306Trp、p.Arg1308Cys、p.Arg1341Gln和p.Val1316Met 4种突变约占2B型突变的90%,其中p.Val1316Met会导致更为严重的血小板减少和出血症状,增加孕期流产风险[14,21]。p.Arg1308Leu和p.Pro1266Gln/Leu为经典的非典型2B型突变,不影响多聚体分布,也不会使血小板减少[20,22]。SACCO等[23]报告了第1例携带D′和D4结构域突变的2B型VWD,并发现p.Arg924Gln/p.Ala2178Ser双杂合突变引起VWF分子的构象转变而导致2B型VWD表型。
2.2.32M型VWD 2M型VWD的发病机制为VWF-HMWMs与血小板GPⅠb或胶原的亲和力减弱,其多聚体分布基本正常。2M型突变为A1区域内的功能缺失性突变,损害VWF与GPⅠb的相互作用使VWF与GPⅠb的结合能力减弱。绝大多数为错义突变,如p.Ser1285Phe、p.Gly1324Ser/Ala、p.Glu1359Lys、p.Phe1369Ile和p.Ile1425Phe,其余部分为小的框内缺失,如p.Lys1408delLys。部分位于A3结构域的错义突变直接降低VWF与胶原结合的亲和力也导致2M型VWD[24],如p.Ser1731Thr、p.Leu1733Pro、p.Ser1738Ala、p.Trp1745Cys、p.Ser1783Ala、p.His1786Asp。2M型VWD一般对DDAVP治疗反应不佳,但A3结构域突变的患者对DDAVP反应良好[18,25]。
2.2.42N型VWD 2N型VWD较为少见,由VWF结合FⅧ的能力缺陷引起,为常染色体隐性遗传,基因型可以是单一2N突变的纯合子、2种不同2N突变的复合杂合子或一种2N突变和一种VWF无效突变的复合杂合子(同时伴有血浆VWF:Ag水平降低)。突变主要位于VWF-FⅧ结合位点D′和部分D3结构域(Ser764-Argl035)内,一些结合区域附近的突变也可以阻碍VWF-FⅧ结合[20],如p.Gln1053His、p.Cys1060Arg。此外,实现VWF-FⅧ结合需要VWFpp从VWF成熟亚单位中裂解,furin酶作用位点在Arg763-Ser764,同时Arg760和Lys762确保furin酶对底物的恰当识别,因此p.Arg760Cys、p.Arg763Gly等突变通过影响furin酶切而抑制VWF-FⅧ结合,导致2N型VWD[20,26]。超过90%的2N型突变为错义突变,其中p.Arg816Trp和p.Arg854Gln最常见。FⅧ活性(FⅧ:C)水平与特定的突变相关,例如,p.Arg816Trp突变导致FⅧ:C严重下降(<10 IU/dL),而p.Arg854Gln突变则导致FⅧ:C水平约为25 IU/dL[20]。大多数2N型VWD患者VWF多聚体分布正常,但部分突变会促使超大型多聚体生成[26],如p.Arg760Cys、p.Arg763Gly、p.Tyr795Cys、p.Gln1053His;部分突变会导致VWF-HMWMs减少[26],如p.Cys788Tyr、p.Cys804Phe、p.Cys1060Arg、p.Asp879Asn。
2.33型VWD 3型VWD以VWF完全缺乏为特征,发病率最低,在所有VWD患者中所占比例<1%,经典遗传模式为常染色体隐性遗传,由纯合或复合杂合突变导致VWF合成或分泌重度缺陷引起。然而据报道,40%~50%的3型VWD患者表现出共显性遗传,其家族中的杂合子携带者符合1型VWD的诊断标准。HGMD数据库中记录了超过320种不同的3型VWD基因突变,无义突变最常见,错义突变次之,其余包括各种剪切突变、缺失/插入、基因转换等,分子致病机制为VWF合成或分泌减少。(1)VWF合成减少。除错义突变外的绝大多数突变(约80%)均产生无效等位基因,且广泛分布于各结构域;而错义突变在D1~D2和CK结构域显示出聚集性,破坏VWFpp构象使VWF多聚化受损或破坏VWF的二聚体化[12],如p.Gly39Arg、p.Asp141Tyr/Asn、p.Lys157Glu、p.Cys275Ser、p.Cys2574Trp、p.Cys2806Arg。(2)VWF分泌减少。部分位于D1~D2结构域的突变阻止VWF从内质网向高尔基体转运的同时抑制WPBs生成,导致VWF内质网滞留、储存和分泌障碍[27],如p.Gly55Glu、p.Val86Glu、p.Trp191Arg和 p.Cys608Trp。
3 VWD的临床诊断
VWD的诊断很复杂,需要出血个人史、出血或VWD的家族史及确认性实验室检测。对于疑似VWD的患者,首先推荐使用国际血栓与止血协会(ISTH)开发的出血评分工具(ISTH-BAT)进行评估,分数异常(男性≥4分,女性≥6分,儿童≥3分)则需完善VWD相关实验室检测。对于转诊到血液内科和(或)一级亲属确诊为VWD的男性和儿童,即使出血评分正常,也应进行进一步的实验室检查。VWD的诊断试验包括筛查、确诊以及分型试验三大部分。筛查试验包括血小板计数(PLT)、活化部分凝血活酶时间(APTT)、凝血酶原时间(PT)及血浆纤维蛋白原(Fg)测定;确诊试验包括VWF:Ag、VWF-血小板结合活性(VWF:Rco,VWF:GPⅠbM,VWF:GPⅠbR)和FⅧ:C测定;分型试验包括VWF多聚体分析、瑞斯托霉素诱导的血小板聚集(RIPA)试验、VWF-胶原结合活性(VWF:CB)、VWF-FⅧ结合活性(VWF:FⅧB)、VWFpp测定、DDAVP试验及VWF基因测序等。
VWF瑞斯托霉素辅因子测定(VWF:Rco)是历年来检测VWF活性的“金标准”,且根据VWF:RCo/VWF:Ag可以确定VWF-血小板结合活性缺陷是由于VWF的质量异常还是数量异常。然而,VWF:Rco具有显著的局限性。一方面,VWF:Rco变异系数高,易出现误诊或漏诊;另一方面,VWF:Rco检测下限为10~20 IU/dL,这使得在低VWF:Ag患者中准确识别2型VWD存在困难[28]。此外,VWF:Rco使用瑞斯托霉素在体外桥接VWF与GPⅠb,所以有可能因为VWF结合瑞斯托霉素的能力缺陷而出现错误结果[28],VWF:Rco明显下降,但实际体内血小板依赖的VWF活性正常。近年来开发出了新的检测方法:功能获得性突变的GPⅠb 结合分析(VWF:GPⅠbM)和瑞斯托霉素诱导的 GPⅠb 结合分析(VWF:GPⅠbR)。VWF:GPⅠbM使用功能增益性突变的GPⅠb,使其在体外不需要瑞斯托霉素也可自发地结合VWF[29]。VWF:GPⅠbM同时还具有高精确度及低变异系数的优势[29],是指南推荐的血小板依赖的VWF活性检测方法[30]。VWF:GPⅠbR使用重组GPⅠb片段而非血小板,受影响因素较少,检测下限更低,但仍需使用瑞斯托霉素。
1型VWD患者血浆VWF水平在3~50 IU/dL,临床表现为轻至中度的皮肤黏膜出血。对VWF:Ag为30~50 IU/dL伴异常出血的患者及无论出血情况如何,VWF:Ag<30 IU/dL的患者,均应考虑1型VWD的诊断,当VWF-血小板结合活性(如VWF:Rco,VWF:GPⅠbM,VWF:GPⅠbR)与VWF:Ag的比值>0.7时,可诊断为1型VWD[30-31]。1C型VWD的特点是VWF半衰期明显缩短至1~3 h(正常8~12 h),VWFpp/VWF:Ag增加至>3.0(正常<3.0)。DDAVP可使储存在血管内皮细胞WPBs中的VWF释放,但对于1C型VWD患者,释放的VWF会很快被清除,DDAVP输注后4 h血浆VWF水平比1 h(峰值)降低超过30%[32]。ISTH优先推荐将DDAVP试验作为1C型VWD的诊断依据[30]。
2型VWD的出血严重程度介于1型和3型之间,常表现为瘀斑、鼻出血、牙龈出血、小伤口持续出血、月经过多及术后出血,其中2A型胃肠道出血多见。2N型主要是外伤后或与手术有关的出血,自发性出血通常不严重,女性月经过多和产后出血常见。当VWF-血小板结合活性(如VWF:Rco,VWF:GPⅠbM,VWF:GPⅠbR)与VWF:Ag的比值<0.7时,应考虑2A、2B或2M型VWD,其中2M型多聚体分布正常,2A和2B型同时伴有VWF-HMWMs缺失。RIPA用于进一步区分2B型与2A型。2B型患者瑞斯托霉素诱导的血小板聚集率增强,低水平的瑞斯托霉素(≤0.7 mg/mL)即可诱导血小板的聚集[33-34],但该试验灵敏度较低。2N型VWD患者血浆VWF:Ag 正常或轻度减少,未结合的FⅧ加速清除导致FⅧ:C减少至5~40 IU/dL,少数患者表现出更明显的下降(1~5 IU/dL),但迄今从未低于1 IU/dL,其诊断标志是VWF:Ag和FⅧ:C水平明显不一致,FⅧ:C/VWF:Ag明显降低。2N型VWD需要与轻型/中间型血友病A及其女性携带者鉴别,通过VWF:FⅧB或基因检测来区分。
3型VWD患者血浆VWF水平通常<3 IU/dL,伴FⅧ:C<10 IU/dL,临床表现为严重的皮肤黏膜出血和关节肌肉出血。
4 VWD的治疗
4.1非替代治疗
4.1.1DDAVP DDAVP可刺激血管内皮细胞分泌储存在WPBs中的VWF,是VWD的有效治疗方法。然而,VWF清除增强的患者在接受DDAVP治疗后血浆VWF水平会迅速降至低水平,DDAVP疗效不佳,特别是在严重出血时。为了充分确定DDAVP治疗后个体VWF的药代动力学反应,建议进行DDAVP试验,即分别检测VWF基线值以及给予DDAVP后1 h、4 h的VWF水平,若VWF增加3倍及以上并可维持则表示对DDAVP反应良好。爱尔兰的一项队列研究表明,大多数VWF水平在30~50 IU/dL的患者在接受DDAVP治疗后有很好的持续性VWF反应,这些患者在诊断时不需要进行正式的DDAVP试验,而是在首次临床应用DDAVP治疗后可以简单地确认VWF反应[29,35]。DDAVP可以皮下注射(0.3 μg/kg)、静脉注射(0.3 μg/kg,溶解于100 mL正常生理盐水中,20 min内输完)或以鼻腔喷雾的形式给药(成年患者300 μg,儿童150 μg)。不良反应包括面部潮红、头痛和低血压,还可引起液体潴留、继发性低钠血症和癫痫发作,建议患者在每次给药后24 h内限制总液体摄入量,高危患者需要监测血钠浓度[28]。2岁以下的儿童以及患有心血管疾病的成人患者通常避免使用DDAVP。2B型VWD患者禁止使用DDAVP,因为释放的VWF可引起明显的血小板减少而加重出血。
4.1.2抗纤维蛋白溶解药物——氨甲环酸(TA) TA通过与纤溶酶原的赖氨酸结合位点结合来抑制纤维蛋白溶解,已被广泛用于治疗VWD,可用于治疗所有类型患者的黏膜出血及大量的月经出血。越来越多的证据强调,即使在高危人群中(如创伤、产后出血、重大骨科手术),TA的使用似乎与显著的血栓风险无关[29]。TA可以口服(15~25 mg/kg,每天3次)、静脉注射(15 mg/kg,每天3次)或作为漱口水使用。不良反应包括恶心、呕吐和腹痛。此外,上尿路有明显血尿的患者一般避免使用TA,因为其可能会导致输尿管血栓性绞痛和梗阻[36]。
4.1.3性激素 对于经期大量出血的无生育需求的女性VWD患者,采用雌激素/孕激素联合避孕药或左炔诺孕酮宫内缓释节育系统能有效地减少经期失血量[37]。
4.2替代治疗
4.2.1血浆源性的VWF浓缩物(pd-VWF) pd-VWF在治疗VWD的出血方面既安全又有效,对于有DDAVP禁忌证的患者,或给予DDAVP后VWF反应不足以应对特定出血事件或手术挑战的患者,pd-VWF是治疗首选。治疗剂量取决于患者的内源性VWF水平和出血的严重程度,剂量标定以制剂的VWF活性为准,由于半衰期约为12 h,通常需要重复给药。大多数pd-VWF同时含有FⅧ,加之患者体内的内源性FⅧ被输注的VWF稳定,因此反复使用pd-VWF可导致血浆FⅧ:C明显升高。FⅧ:C升高已被证明是普通人群中静脉血栓栓塞的一种剂量依赖性危险因素,一些VWD患者在使用pd-VWF后的确出现了血栓并发症,但目前FⅧ:C的升高在pd-VWF治疗后VWD患者血栓形成病因中的重要性尚不清楚[29,36]。VWF大量缺失的患者在重复给予pd-VWF后抑制物形成风险较大,有5%~10%的3型VWD患者接受pd-VWF治疗后出现了抗VWF中和抗体[38]。
4.2.2重组人VWF浓缩物(rVWF) 第一种rVWF已经在许多国家开发并获批用于治疗成人VWD,其在生产过程中不暴露于ADAMTS13,因此富含VWF-HMWMs,对VWD患者的出血疗效较好。此外,rVWF的耐受性强,尽管富含VWF-HMWMs,但无发生血栓或微血管病变并发症的证据,这可能与体内ADAMTS13介导的蛋白分解在rVWF输液后迅速发生相关[39-40]。最重要的是,rVWF不会导致抑制物形成。需注意的是rVWF不含FⅧ,故对伴有FⅧ:C降低的VWD出血患者,在首次用药时需同时补充重组FⅧ(rFⅧ),以确保立即达到止血FⅧ水平。不联合应用rFⅧ的情况下,单独输注rVWF仍可使血浆FⅧ:C正常化,这是由于rVWF稳定了内源性分泌的FⅧ。例如,在单独给药rVWF 6 h后3型VWD患者的FⅧ:C上升至>40 IU/dL,48 h后血浆FⅧ:C仍可维持在>70 IU/dL[41]。与pd-VWF相比,rVWF似乎能更有效地稳定内源性FⅧ,这可能与其更长的半衰期(约25.5 h)或更高的VWF-HMWMs水平有关。
4.2.3血小板源性的VWF 正常富血小板血浆中,VWF总量的15%~20%储存在血小板α颗粒中,在血管损伤部位血小板激活后,以高水平局部分泌,发挥重要的止血作用。有研究表明,血小板源性的VWF不仅富含HMWMs,而且对ADAMTS13蛋白裂解具有部分抗性[29]。未来需要进一步的研究来充分确定血小板源性的VWF的生物学重要性,特别是其在与VWD相关的可变出血表型中的作用。
5 结 语
近年来对VWD分子致病机制的认识取得了重大进展,这些新颖的见解改进了VWD的诊断和治疗策略。新的血小板依赖性VWF活性测定、DDAVP试验、VWF基因测序等手段提高了VWD诊断的准确性,但国内只有极少数医院常规开展;rVWF的面世给VWD的治疗带来了突破,但暂未能被大范围使用。因此,VWD的分型诊断和治疗仍面临困难与挑战,需要更多的工作来简化诊断,优化治疗。
