结合基因关联和转录组分析鉴定小麦成株期抗条锈病位点YrZ501-2BL的候选基因
2023-06-27张旭韩金妤李晨晨张丹丹吴启蒙刘胜杰焦韩轩黄硕李春莲王长发曾庆东康振生韩德俊吴建辉
张旭,韩金妤,李晨晨,张丹丹,吴启蒙,刘胜杰,焦韩轩,黄硕,李春莲,王长发,曾庆东,康振生,韩德俊,吴建辉
结合基因关联和转录组分析鉴定小麦成株期抗条锈病位点的候选基因

1西北农林科技大学农学院/旱区作物逆境生物学国家重点实验室,陕西杨凌 712100;2西北农林科技大学植物保护学院/旱区作物逆境生物学国家重点实验室,陕西杨凌 712100
【目的】小麦条锈病是小麦的主要病害之一,每年都会对小麦产量安全造成严重危害,挖掘小麦抗条锈病基因,为小麦抗条锈病种质创新和揭示小麦抗条锈病遗传机制奠定基础。【方法】利用多组学手段结合全基因关联分析(GWAS)开展对小麦成株期抗条锈病性状的解析。首先对411份来自CIMMYT和ICARDA的春小麦进行全基因组关联分析,在小麦2BL染色体上定位到一个主效的成株期抗条锈病位点,并利用含有该位点的抗病材料Z501及感病亲本晋麦79的双亲群体进行连锁作图,成功验证了该位点抗性的稳定性,暂命名为。在此基础上,通过基因注释、比较基因组分析、转录组分析和候选基因的关联分析对目标区间筛选候选基因。【结果】综合GWAS和连锁作图结果,将锁定在小麦2B染色体0.26 Mb(575.706—576.587 Mb)范围内,根据中国春参考基因组注释信息分析,该区间含有12个基因,其中,高可信基因6个;利用在线网站,将目标区间所在的中国春参考基因组与其他已公布的不同倍性小麦基因组进行比较,发现该区间的6个高可信小麦基因基本都能在其他小麦材料中找到同源基因,且基因排列顺序相同,说明该区间可能不存在较大片段的插入、缺失、倒位等现象,可以利用参考基因组信息进行候选基因预测;结合抗病亲本Z501和感病亲本晋麦79的成株期接种条锈菌后的转录组数据,发现只有、和受诱导表达,且抗感病亲本存在表达差异。根据中国春基因组注释,三者分别编码GATA转录因子、SH3P2蛋白和锌指蛋白。通过进一步的候选基因关联分析,发现只有SH3P2蛋白中存在与条锈病表型显著性差异的SNP位点(G/A),虽然该位点(G1369A)在2个可变剪切的转录本中均未引起氨基酸编码变化(TCG和TCA均编码丝氨酸),但可能与可变剪切有关,同时该位点(G1369A)的不同单倍型表型之间也存在极显著差异,进一步分析发现,在G1369A位点下游还有2个引起氨基酸改变的变异位点G1377A和G1431A,分别引起缬氨酸(GTT)到异亮氨酸(ATT)和缬氨酸(GTG)到甲硫氨酸(ATG)的改变,但这两个位点在455份重测序材料中所占比例只有0.87%,属于稀有变异,因此,未进行显著性检验。综上,推测为的重要抗病候选基因;此外,针对候选区间的差异SNP开发了相应的AQP标记,可用于辅助选择,为下一步小麦抗锈病分子育种应用提供了标记资源。【结论】利用多组学整合关联分析的方法,成功在小麦2B染色体上挖掘到1个抗条锈病候选基因。
小麦;条锈病抗性;;多组学整合分析;SH3P2蛋白
0 引言
【研究意义】小麦条锈病是世界性流行的活体寄生真菌病害,一直是威胁中国小麦安全生产的重要病害。近年来产生的新毒性小种条中34大面积流行,导致当前中国有效的抗条锈病资源极度缺乏,是抗病育种面临的重要问题[1-2]。因此,快速高效地挖掘小麦重要广谱抗性资源,克隆其关键抗病基因是突破这一瓶颈的前提条件[3]。【前人研究进展】随着小麦条锈病致病机理的不断深入探究,发现成株期抗病性往往具有持久性特点,目前,克隆到的成株期抗病基因(如、和)均具有广谱、抗多种病害及持久的特点[4]。其中,编码ABC转运蛋白(ATP binding cassette transporter),通过调控磷脂代谢影响细胞膜结构以及脱落酸信号通路来影响病原菌的生长,达到抗病的目的[5-6];编码一个包含激酶和脂绑定结构域的蛋白(START-kinase),在相对高温(25—35℃)条件下,表现出对多个条锈菌小种的广谱抗性[7-8];编码己糖转运蛋白(hexose transporter),通过位点突变产生显性负效应作用,引起Yr46蛋白丧失对葡萄糖的转运活性,导致入侵的病原菌不能获得足够的糖源,从而使植物表现为抗病[9]。克隆的这3个小麦成株抗性基因,虽抗病机制不同,但均参与能量代谢或糖转运,与衰老有关,且相对保守[4],3个基因在中国春基因组中均具有相对完整的基因结构。受此启发,通过对中国春基因组目标区间内基因进行解析,可能会发现与成株期抗条锈病相关的候选基因[10]。随着小麦参考基因组精细图谱组装的完成以及二代测序、组装和捕获测序的快速发展[11],相继开发出一系列基因克隆的新策略,加快了传统的基因克隆进程[12]。此外,前人开展了大量的全基因组关联分析(genome-wide association study,GWAS)模型、方法的探索,如日本学者通过全基因组测序,在水稻中开发了一种高效GWAS分析方法,基于对每个核苷酸多态性的功能重要性进行评估,可以快速鉴定候选基因,而不需要额外的试验[13]。同样,在小麦中也开展了类似研究,如利用抗病R基因富集测序结合关联分析开展的小麦秆锈病R基因快速克隆[14],利用外显子捕获测序结合关联分析快速检测小麦抗叶锈病基因的功能SNP变异[15],以及利用高密度SNP芯片结合关联分析开展候选基因的发掘与功能验证等[16-19],使研究者能够利用多组学手段结合GWAS开展对小麦复杂性状的解析。国际玉米小麦改良中心(CIMMYT)和国际干旱农业研究中心(ICARDA)的春小麦材料对小麦条锈病具有良好的抗性,前期通过大规模基因组关联分析在小麦2BL染色体快速发掘4个抗条锈病QTL位点[10],其中,和均已被成功验证[10, 20]。【本研究切入点】虽然部分位点已经过验证,但其他兴趣目标区间(如第三个主效位点)遗传基础还不清楚,特别是与抗条锈病有关的候选基因分析方面,尚未进行深入解析。【拟解决的关键问题】本研究着眼于第三个主效位点,利用连锁作图、候选基因关联分析、转录组分析等多种手段对目标区间内的候选基因进行快速筛查,确定与抗条锈病有关的关键候选基因,为小麦抗条锈病种质创新和揭示小麦抗条锈病遗传机制奠定基础。
1 材料与方法
1.1 试验材料和遗传群体创制
全基因组关联分析材料:实验室前期收集了411份主要来自国际玉米小麦改良中心(CIMMYT)和国际干旱农业研究中心(ICARDA)的春小麦高代品系材料(电子附表1),其中202份ICARDA材料由西北农林科技大学农学院宋卫宁教授惠赠[21],该套材料已完成了在中国条锈病常发区的多年多点的条锈病表型鉴定[21],并进行小麦660K芯片的基因分型,进一步完成了小麦条锈病的GWAS定位工作,在小麦染色体2BL上定位多个抗病QTL,其中包含[10]。
连锁分析:通过GWAS分析,上述411份小麦材料中,来自澳大利亚抗病品种Z501(PI 410902,系谱为IRN-62-101/Cheyenne)可能含有目标位点,为验证该位点的准确性及稳定性,以感病品种晋麦79作为受体母本,Z501为供体父本,杂交构建306个F2和F2:3遗传分析群体,并构建连锁图谱进行基因定位。
候选基因关联分析:实验室前期收集了455份已进行基因组重测序的材料(其中绝大部分已公开发表)[22-24],该套材料包含农家种、中国历年审定品种、国外引进品种,以及高代品系等,部分材料由山西农业大学郑军副教授惠赠。关于该套材料的详细信息请见电子附表1。
感病对照品种为铭贤169(MX169)和小偃22(XY22)。
1.2 全基因组关联分析
利用GEMMA软件中的单变量线性混合模型对411份小麦材料的成株期表型(实验室已有多年多点表型数据)及基因型进行全基因组关联分析[10]。其中,e表示用GEC(Genetic Type I Error Calculator)软件[25]计算的有效SNP位点数目。利用GEC软件计算各染色体建议值的范围为1.36×10-4—9.9×10-4,所以整体考虑3.40×10-4作为显著性位点的阈值。选择R软件的QQman软件包进行绘制曼哈顿图[26],采用GCTA软件[27]计算位点染色体的遗传率。
1.3 田间试验及遗传分析
2017—2018年和2018—2019年播种季在陕西省杨凌西北农林科技大学试验田种植亲本及306个F2单株和F2:3家系。F2:3家系每个家系行长1 m,行距0.2 m,每隔20个家系种植2行小偃22作为感病对照,周边种植铭贤169作为诱发行。试验田采取人工接种手段,使用的小麦条锈菌为CYR32、CYR33、CYR34(1﹕1﹕1)。
当感病对照品种铭贤169和小偃22叶片发病面积>80%时,开始对遗传群体进行表型鉴定。每隔3 d重复一次鉴定,共调查3次,采用反应型(infection type,IT)[28]为鉴定标准,取最高反应型为最终抗病性结果。对于纯合抗病/感病家系,只记录一个表型数值;对于表型分离家系,记录2个表型数值,即最小值和最大值。在对该群体进行遗传分析时,利用Excel软件进行统计分析,参考IT数据,其中,0—3为高抗反应型,4—6为中抗反应型,7—9为感病反应型[29]。
1.4 混池测序(BSA)与基因分型
根据F2和F2:3家系抗条锈病鉴定结果,挑选20个纯合抗病家系(IT=0—3)和20个纯合感病家系(IT=9)构建抗感池,将双亲与抗感池送至北京博奥生物技术有限公司进行小麦660K SNP芯片的全基因组扫描,分析在抗感池和双亲之间多态性SNP位点在染色体上的分布。利用Excel软件IF函数筛选抗病亲本、感病亲本以及抗感池之间的纯合差异SNP位点,并参照整合的遗传图谱获得差异SNP位点的染色体分布和物理位置信息[18]。随后,使用在线设计平台网站PolyMarker(http://www.polymarker.info/)对所有纯合位点的SNP进行等位基因特异的定量PCR基因分型系统(allele-specific quantitative PCR based genotyping assay,AQP)标记[30]设计,再挑选出染色体特异性的标记,根据SNP的物理参考位置及分布密度选取若干AQP标记送公司合成引物。用亲本、抗感池和随机挑选的小样本群体验证标记的特异性,将筛选合格的多态性标记用于作图群体进行基因分型。
1.5 遗传连锁图谱构建及基因定位
各群体的分离比例通过卡方(χ2)分析确定实际值与期望值的显著性,利用JOINMAP 4.0软件[31]进行分子标记与条锈病表型之间的连锁分析,LOD值设为3.0。采用Kosambi函数[32]计算遗传距离。采用软件Mapchart V2.3[33]绘制遗传图谱。
1.6 比较基因组分析
根据定位结果确定目标位点两翼最近的标记在中国春参考基因组(IWGSC RefSeqv 1.1)[34]对应的物理位置,在开放网站http://wheat.cau.edu.cn/TGT/上[35],利用10+Genome等基因组信息,进行比较基因组分析,用以观察目标区间的共线性、染色体结构是否发生变异等情况。
1.7 候选基因预测
目前,小麦族多组学网站(Wheatomics,http://wheatomics.sdau.edu.cn/)[36]整合了已发表的多个小麦族物种的基因组、转录组、变异组等多组学数据。根据GWAS和遗传定位的整合结果,在该网站中获取目标区间两端最近的侧翼标记之间的中国春IWGSC RefSeq v1.1参考基因组序列,并根据区间内部指定基因的ID获取对应的基因序列,结合实验室前期针对Z501和晋麦79的转录组数据,利用基因组浏览器(JBrowse)对目标区间所有鉴定到的基因进行以下3个层面的分析:1)表达的时空特异性,比较候选基因在不同发育时期、不同材料之间的表达差异;2)mRNA水平是否存在多态性,主要比较候选基因在感病和抗病材料之间是否存在序列变异,这些变异是否导致无义、错义、移码突变或剪切模式的改变;3)表达量的变化,利用RNA-Seq数据确认候选基因在不同材料或组织中表达量的相对差异。
1.8 候选基因的关联分析
调取候选基因在455份普通小麦上的重测序数据,所得序列需覆盖基因的外显子、内含子、5′-和3′-非翻译区、启动子区。通过与参考基因组中国春序列进行比对,分析核苷酸多态性,包括SNP和InDels。在此基础上,结合小麦成株期抗条锈病表型,应用GAPIT软件[37]进行关联分析,鉴定与抗病表型显著关联的功能性遗传变异及抗病优异单倍型,并绘制LD-map。
针对候选区域内,基于以下原则对核酸多样性进行分类[13]:G1:引起氨基酸编码变化和可变剪切位点的显著关联SNP;G2:位于启动子区(起始密码子ATG上游2 kb)内显著关联SNP;G3:位于编码区的同义突变,内含子区、3′ UTR区显著关联的SNP;G4:编码区(coding regions)外的显著关联SNP;G5:不显著关联的SNP。
2 结果
2.1 全基因组关联分析
在单变量线性混合模型分析的基础上,对411份材料在多年多个田间环境下的条锈病表型(IT和DS值)和最佳线性无偏预测(BLUP)进行关联检验[10]。为方便表述,本文只列出DS-BLUP的GWAS结果(图1-a),在2B染色体长臂上发现极为显著的峰值,进一步分析,在该2BL上共发现4个QTL位点,其中,和均在前期已被本课题组验证[10, 20]。本研究着眼于第3个主效位点(图1-b),通过整合多个环境的GWAS结果,发现绝大部分显著性SNP位点都集中在575.706—576.587 Mb范围内,因此,推断该区间为的重要目标区间(图1-c和图1-g)。

a:利用411份春小麦品系进行条锈病表型GWAS分析,全基因组显著阈值-log10(P)为3.4(c同理);b、c:2B染色体峰周围的基于单核苷酸多态性关联的曼哈顿局部图,灰色虚线包围的区域代表潜在的候选区域;d:BSA+660K SNP芯片中差异的多态SNPs在小麦每条染色体上的分布情况;e:多态SNP在2B染色体上的物理位置分布密度;f:基于基因型数据的小麦2B染色体上QYr.nwafu-2BL.1(YrZ501)位点的遗传连锁图谱,红色条表示QYr.nwafu-2BL.1(YrZ501)位点的候选区间;g:QYr.nwafu-2BL.1(YrZ501)候选区间内的预测基因
2.2 晋麦79×Z501的群体抗病遗传分析
为进一步确认抗病位点的准确性,根据GWAS结果,找到含有该位点的小麦材料Z501,并对晋麦79×Z501的亲本及其后代F2植株和F2:3家系进行表型统计分析。抗病亲本Z501表现为高抗(IT=1—3),感病亲本晋麦79表现为高感(IT=9)(图2)。根据F2单株的反应型看,抗病单株67个,感病单株239个,经卡方测验(χ2=1.57,=0.20>0.05),符合1﹕3期望比,从F2:3家系的反应型看,纯合抗病家系数有67个,纯合感病家系数有77个,抗感分离家系数150个,经卡方测验(χ2=1.71,=0.42>0.05),符合1﹕2﹕1期望比(表1),说明小麦抗病材料Z501的抗性由1对主效基因控制。
2.3 BSA与基因定位
根据BSA结果,在抗、感池之间共有2 057个差异SNP位点,依据小麦660K SNP的染色体参考位置,其中,2B染色体上有904个差异SNP,剩余的差异SNP分布于其他染色体上,而且在2B染色体上抗感池差异性位点与亲本间差异性位点重叠比例最高(图1-d)。按照每1 Mb统计SNP差异位点数目,发现2B上的差异SNP大部分在500—600 Mb(染色体总长度为800 Mb)区间内(图1-e)。为了确定目标基因所在区间,在区间500—600 Mb内开发了76个染色体特异性AQP标记,经过亲本、抗感池以及小群体验证,其中24个标记特异性良好,并在F2群体上进行基因分型,将标记检测的基因型数据和表型数据进行整合,利用Joinmap软件绘制出全长22.7 cM的遗传图谱,最终将目标基因锁定在AQP标记和间隔1.1 cM区间上(图1-f),对应物理区间为572.628—577.452 Mb(图1-g),与全基因组关联分析锁定的区间范围基本一致,对该位点暂命名为。
2.4 比较基因组分析
调取目标区域在中国春参考基因组中物理区间(575.706—576.587 Mb)内所有高可信度基因,分别与10+Genome等不同六倍体小麦品种基因组以及四倍体基因组进行比对分析。目标区域在中国春小麦参考基因组上跨度为0.26 Mb,对应的其他六倍体基因组区间在0.26—0.65 Mb不等,在六倍体斯卑尔脱小麦、四倍体硬粒小麦、野生二粒小麦和拟斯卑尔脱山羊草跨度在0.27—0.66 Mb不等(图3),说明目标区域在从二倍体到六倍体小麦演化过程中发生了片段的插入缺失。进一步分析发现,该区间的6个高可信度小麦基因,基本都能在其他小麦材料中找到同源基因(图3),且基因排列顺序相同,极个别基因组存在某个基因的丢失和插入现象,但整体共线性良好,可以利用参考基因组信息进行候选基因预测分析。
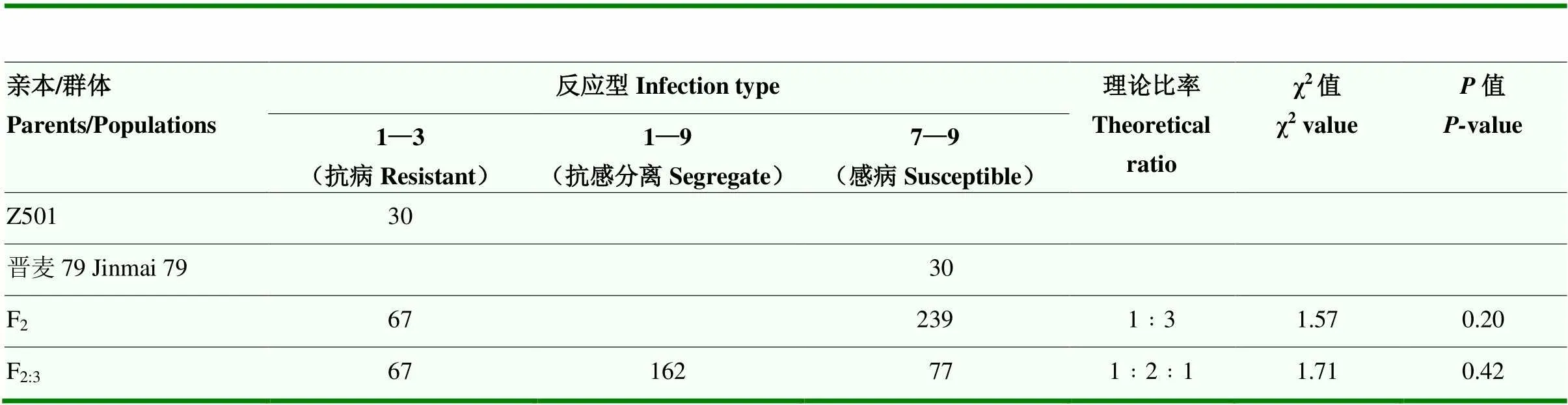
表1 晋麦79×Z501群体田间抗性反应统计及遗传分析

图2 小麦材料Z501和晋麦79田间抗病表现

图3 YrZ501物理区间及其与不同倍性小麦材料基因组的共线性分析
2.5 候选基因预测
根据中国春参考基因组的注释信息,在的定位区间内共含有12个基因(图4-a),根据Z501和晋麦79成株期接种条锈菌后的6个时间点(0、1、2、4、7和14 d)转录表达量分析(图4-b),6个低可信基因均未表达,也没有表达,和在接种前后表达量差异不显著,、和在接种前后的表达量差异显著,且抗病Z501和感病晋麦79之间也存在差异。其中,编码GATA转录因子;属于的一种可变剪切,编码SH3P2蛋白;、分别是的2种可变剪切,均编码B-box锌指结构蛋白。根据前人的研究,三者均与抗病有关,因此,推断、和可能是的候选基因。

a:候选区间内基因功能注释信息;b:亲本Z501与晋麦79的转录组表达量
2.6 候选基因关联分析
针对2.5中初步选定的3个候选基因,调取其在455份小麦重测序的序列变异,并进行候选基因关联分析,取-log10()=3阈值,结果显示,基因和中均未发现显著变异SNP位点(图5-a—b),仅在中有一个显著性变异SNP位点(G1369A)(图5-c),抗病亲本Z501在该位点处与中国春参考基因组相同,均为G,而感病亲本晋麦79在该位点处发生了变异,由G变为A,根据1.8中差异SNP分类原则,该位点(G1369A)在2个可变剪切的转录本中均未引起氨基酸编码变化(TCG和TCA均编码丝氨酸)(图5-d),但可能与可变剪切有关,同时,该位点(G1369A)的不同单倍型表型之间也存在极显著差异(图5-e)。进一步分析发现,在G1369A位点下游还有2个引起氨基酸改变的变异位点——G1377A和G1431A,分别引起缬氨酸(GTT)到异亮氨酸(ATT)和缬氨酸(GTG)到甲硫氨酸(ATG)的改变(图5-d),但2个位点在455份重测序材料中所占比例只有0.87%,属于稀有变异,在利用GAPIT分析时被过滤掉,因此,未进行显著性检验。综上,推测的可变剪切转录本为的重要抗病候选基因。
3 讨论
3.1 关联分析与连锁作图相结合可有效解析复杂性状
近年来,关联分析和连锁作图相结合来解析复杂数量性状已成为越来越重要的方法。关联分析以数百个种质组成的自然群体为研究材料,由于历史重组事件的积累而产生的大量遗传变异,可以显著地增加变异范围,而且可以同时检测同一基因座上的多个等位基因[38]。然而,关联分析存在2个缺陷,一是关联分析无法检测稀有等位变异;二是当自然群体存在较为复杂的群体结构时,可能导致基因多态性位点与性状的相关性并非由功能性等位基因引起,从而出现假阳性结果[38]。与之相比,连锁作图已被证明可以有效地剖析并定位复杂性状的基因/QTL,但由于所应用的亲本数较少,后代群体重组的机会有限,因此,当等位基因在双亲中无差异时,基因/QTL无法通过连锁作图进一步实现精细定位[39]。不论是连锁作图,还是关联分析在进行QTL检测时均具有一定的优缺点,所以充分利用2种方法的优势,将2种方法相结合不但能够显著提高基因/QTL的检测效率,还能有助于发现可靠、稳定的基因/QTL。例如Zhang等[40]利用228个重组自交系定位到与黄曲霉毒素积累显著相关的QTL(),随后又用437份自交系和558 629个SNP标记对黄曲霉毒素进行全基因组关联分析,鉴定出了25个显著的SNP位点,且这些SNP位点大部分与共定位,最终将缩小到1.5 kb区间内;常立国等[41]利用150份重组自交系群体和139份自然材料组成的关联群体同时对持绿相关性状进行定位,共同检测到4个遗传稳定的共定位遗传区段;ZhaNG等[42]利用375份油菜自然群体和150个DH遗传群体,通过关联和连锁分析共定位到一个新的控制油酸含量的主效QTL,并将其精细定位在76 kb的区间;刘凯等[43]利用小麦重组自交系群体和由205个品种(系)构成的自然群体,对两群体的茎秆断裂强度相关性状进行连锁作图和全基因组关联分析。在4B染色体上,检测到9个与小麦茎秆断裂强度相关QTL,最后定位到6.7 cM区间内。课题组前期利用411份CIMMYT和ICARDA春小麦材料通过关联分析在2BL染色体上定位出大量的主效稳定的基因/QTL,并利用遗传群体分别确认了和的可靠性。本试验通过晋麦79×Z501遗传群体的连锁作图验证了另外一个GWAS主效位点,为快速发掘有效抗条锈病基因/QTL资源提供了有效手段。
3.2 多组学研究手段结合新方法是实现抗病基因快速克隆的有效途径
随着测序技术不断更新迭代以及成本的下降,海量数据呈井喷式涌现出来。使得科研工作者可以采用大数据分析、多组学研究手段以及多种新方法结合分析,迅速克隆目的基因并对其调控机理进行全面解析。如,利用3 000多份水稻重测序数据,结合全基因组关联分析快速挖掘了水稻广谱抗稻瘟病遗传位点[44];整合分析番茄转录组和代谢组,迅速锁定了法卡林二醇的生物合成基因簇参与番茄对病害胁迫的反应[45];利用RNA深度测序发现miRNA参与植物的抗病性,并揭示了多个miRNA参与病害调控的机理[46];采用结构生物学的手段解析了抗病小体结构,揭示了NLR蛋白调控免疫的新机理等[47]。近些年,小麦在多组学上也取得了一系列突破和进展,截至目前,二倍体、四倍体、六倍体小麦的基因组精细图谱相继完成[48],其他六倍体小麦品种基因组测序和组装,如国外10+Pan-genome[49],中国自主测定组装科农9204等也陆续释放[50],此外,小麦的大规模重测序、外显子测序、全生育期转录组、代谢组工作也如火如荼开展起来[19, 21, 23, 51-53]。这些海量的基因组数据提供了丰富的参考信息和技术手段,极大促进了对小麦抗病基因挖掘。例如,突变体R基因富集测序(mutant R gene enrichment sequencing,MutRenSeq)、突变体目标染色体测序(mutant chromosome sequencing,MutChromSeq)、目标染色体长片段组装(targeted chromosome-based cloning via long-range assembly,TACCA)等技术体系的提出,与传统的图位克隆方法相比,无需构建大规模的遗传作图群体,无需构建BAC文库,不完全依赖参考基因组,对基因的克隆快速、高效、准确[54];近年来衍生出来的利用关联分析结合R基因富集测序的方法(AgRenSeq),可以快速同时实现对多个抗秆锈病基因的克隆[14];利用高密度SNP芯片、重测序等基因组大数据通过关联分析和连锁作图,并利用转录组、变异组等信息基于每个核苷酸多态性的功能重要性进行评估,快速开展了小麦成株期抗条锈病候选基因的克隆[10]和抗茎基腐病候选基因的克隆[55]等工作。充分利用当前小麦基因组学及功能基因组学最新的研究成果,整合现有的技术和方法,相互补充和完善,是发掘和克隆重要基因的一条有效途径。本研究同样整合了多组学手段,综合分析快速发掘出的候选基因,为进一步克隆并验证其功能奠定了坚实基础。
3.3 YrZ501与2BL染色体上其他抗病位点的关系及潜在的育种价值
来源于春小麦Z501,被定位在IWGSC RefSeq v1.1参考基因组2BL染色体的575.706—576.587 Mb约119 kb范围。根据前人对于小麦抗病QTL元分析的结果[1, 56],在2BL染色体区段的310—783 Mb(几乎整个染色体长臂)都有大量的抗病QTL定位出来,其中,包含已正式命名的基因、/、、和,这些基因都认为是全生育期抗性基因,目前,除了和外,其他基因对中国当前的条锈菌流行小种基本失效[1],且和分别来自斯卑尔脱小麦(ssp.var.)和硬粒小麦(),因此,从抗性类型、来源等推断,与二者不同。前人在该染色体区段已定位出多个成株期抗病QTL,包括CIMMYT春小麦的、美国冬小麦Druchamp的-、德国春小麦Naxos的-、意大利冬小麦Aquileja的-、法国冬小麦Camp Remy的-、中国冬小麦陕麦155的、中国冬小麦秦农142的-等[1, 56]。与以上部分位点有重叠,很可能为同一个抗性位点,但需要进一步的等位性测试。除了和-,其他位点基本都处于粗定位阶段,存在定位精确度低、区间大等问题,极大限制了其在小麦抗病育种中的应用。本研究针对候选区间的差异SNP开发了相应的AQP标记,可用于辅助选择,为下一步小麦抗锈病分子育种应用提供了标记资源。
4 结论
结合关联分析和连锁作图在小麦2BL染色体上共定位到一个抗条锈病主效基因,并通过基因功能注释、转录组数据分析、比较基因组分析、候选基因关联分析预测为重要候选基因。
[1] 韩德俊, 康振生. 中国小麦品种抗条锈病现状及存在问题与对策. 植物保护, 2018, 44(5): 1-12.
HAN D J, KANG Z S. Current status and future strategy in breeding wheat for resistance to stripe rust in China. Plant Protection, 2018, 44(5): 1-12. (in Chinese)
[2] 康振生, 王晓杰, 赵杰, 汤春蕾, 黄丽丽. 小麦条锈菌致病性及其变异研究进展. 中国农业科学, 2015, 48(17): 3439-3453.
KANG Z S, WANG X J, ZHAO J, TANG C L, HUANG L L. Advances in research of progress evariation of the wheat stripe rust fungusf. sp.. Scientia Agricultura Sinica, 2015, 48(17): 3439-3453. (in Chinese)
[3] 邓一文, 刘裕强, 王静, 陈学伟, 何祖华. 农作物抗病虫研究的战略思考. 中国科学: 生命科学, 2021, 51(10): 1435-1446.
DENG Y W, LIU Y Q, WANG J, CHEN X W, HE Z H. Strategic thinking and research on crop diseases and pest resistance in China. Scientia Sinica (Vitae), 2021, 51(10): 1435-1446. (in Chinese)
[4] KOURELIS J, van der HOORN R A L. Defended to the nines: 25 years of resistance gene cloning identifies nine mechanisms for R protein function. The Plant Cell, 2018, 30(2): 285-299.
[5] KRATTINGER S G, LAGUDAH E S, SPIELMEYER W, SINGH R P, HUERTA-ESPINO J, MCFADDEN H, BOSSOLINI E, SELTER L L, KELLER B. A putative ABC transporter confers durable resistance to multiple fungal pathogens in wheat. Science, 2009, 323(5919): 1360-1363.
[6] KRATTINGER S G, KANG J, BRÄUNLICH S, BONI R, CHAUHAN H, SELTER L L, ROBINSON M D, SCHMID M W, WIEDERHOLD E, HENSEL G, KUMLEHN J, SUCHER J, MARTINOIA E, KELLER B. Abscisic acid is a substrate of the ABC transporter encoded by the durable wheat disease resistance gene Lr34. New Phytologist, 2019, 223(2): 853-866.
[7] FU D L, UAUY C, DISTELFELD A, BLECHL A, EPSTEIN L, CHEN X M, SELA H N, FAHIMA T, DUBCOVSKY J. A kinase-START gene confers temperature-dependent resistance to wheat stripe rust. Science, 2009, 323(5919): 1357-1360.
[8] GOU J Y, LI K, WU K T, WANG X D, LIN H Q, CANTU D, UAUY C, DOBON-ALONSO A, MIDORIKAWA T, INOUE K, SÁNCHEZ J, FU D L, BLECHL A, WALLINGTON E, FAHIMA T, MEETA M, EPSTEIN L, DUBCOVSKY J. Wheat stripe rust resistance protein WKS1 reduces the ability of the thylakoid-associated ascorbate peroxidase to detoxify reactive oxygen species. The Plant Cell, 2015, 27(6): 1755-1770.
[9] MOORE J W, HERRERA-FOESSEL S, LAN C X, SCHNIPPENKOETTER W, AYLIFFE M, HUERTA-ESPINO J, LILLEMO M, VICCARS L, MILNE R, PERIYANNAN S, KONG X Y, SPIELMEYER W, TALBOT M, BARIANA H, PATRICK J W, DODDS P, SINGH R, LAGUDAH E. A recently evolved hexose transporter variant confers resistance to multiple pathogens in wheat. Nature Genetics, 2015, 47(12): 1494-1498.
[10] WU J H, YU R, WANG H Y, ZHOU C E, HUANG S, JIAO H X, YU S Z, NIE X J, WANG Q L, LIU S J, SONG W N, SINGH R P, BHAVANI S, KANG Z S, HAN D J, ZENG Q D. A large-scale genomic association analysis identifies the candidate causal genes conferring stripe rust resistance under multiple field environments. Plant Biotechnology Journal, 2021, 19(1): 177-191.
[11] BEVAN M W, UAUY C, WULFF B B H, ZHOU J, KRASILEVA K, CLARK M D. Genomic innovation for crop improvement. Nature, 2017, 543(7645): 346-354.
[12] BETTGENHAEUSER J, KRATTINGER S G. Rapid gene cloning in cereals. Theoretical and Applied Genetics,2019, 132(3): 699-711.
[13] YANO K, YAMAMOTO E, AYA K, TAKEUCHI H, LO P C, HU L, YAMASAKI M, YOSHIDA S, KITANO H, HIRANO K, MATSUOKA M. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nature Genetics, 2016, 48(8): 927-934.
[14] ARORA S, STEUERNAGEL B, GAURAV K, CHANDRAMOHAN S, LONG Y M, MATNY O, JOHNSON R, ENK J, PERIYANNAN S, SINGH N, ASYRAF MD HATTA M, ATHIYANNAN N, CHEEMA J, YU G T, KANGARA N, GHOSH S, SZABO L J, POLAND J, BARIANA H, JONES J D G, BENTLEY A R, AYLIFFE M, OLSON E, XU S S, STEFFENSON B J, LAGUDAH E, WULFF B B H. Resistance gene cloning from a wild crop relative by sequence capture and association genetics. Nature Biotechnology, 2019, 37(2): 139-143.
[15] LIU F, ZHAO Y S, BEIER S, JIANG Y, THORWARTH P, LONGIN C F H, GANAL M, HIMMELBACH A, REIF J C, SCHULTHESS A W. Exome association analysis sheds light onto leaf rust () resistance genes currently used in wheat breeding (L.). Plant Biotechnology Journal, 2020, 18(6): 1396-1408.
[16] GUO Z F, CHEN D J, ALQUDAH A M, RÖDER M S, GANAL M W, SCHNURBUSCH T. Genome-wide association analyses of 54 traits identified multiple loci for the determination of floret fertility in wheat. The New Phytologist, 2017, 214(1): 257-270.
[17] LI L, MAO X G, WANG J Y, CHANG X P, REYNOLDS M, JING R L. Genetic dissection of drought and heat-responsive agronomic traits in wheat. Plant, Cell & Environment, 2019, 42(9): 2540-2553.
[18] SUN C W, DONG Z D, ZHAO L, REN Y, ZHANG N, CHEN F. The Wheat 660K SNP array demonstrates great potential for marker- assisted selection in polyploid wheat. Plant Biotechnology Journal, 2020, 18(6): 1354-1360.
[19] CHEN J, HU X, SHI T T, YIN H R, SUN D F, HAO Y F, XIA X C, LUO J, FERNIE A R, HE Z H, CHEN W. Metabolite-based genome-wide association study enables dissection of the flavonoid decoration pathway of wheat kernels. Plant Biotechnology Journal, 2020, 18(8): 1722-1735.
[20] ZHOU C E, LIU D, ZHANG X, WU Q M, LIU S J, ZENG Q D, WANG Q L, WANG C F, LI C L, SINGH R P, BHAVANI S, KANG Z S, HAN D J, ZHENG W J, WU J H. Combined linkage and association mapping reveals two major QTL for stripe rust adult plant resistance in Shaanmai 155 and their haplotype variation in common wheat germplasm. The Crop Journal, 2022, 10(3): 783-792.
[21] WANG T T, SU N, LU J N, ZHANG R P, SUN X M, SONG W N. Genome-wide association studies of peduncle length in wheat under rain-fed and irrigating field conditions. Journal of Plant Physiology, 2023, 280: 153854.
[22] GUO W L, XIN M M, WANG Z H, YAO Y Y, HU Z R, SONG W J, YU K H, CHEN Y M, WANG X B, GUAN P F, APPELS R, PENG H R, NI Z F, SUN Q X. Origin and adaptation to high altitude of Tibetan semi-wild wheat. Nature Communications, 2020, 11: 5085.
[23] ZHOU Y, ZHAO X B, LI Y W, XU J, BI A Y, KANG L P, XU D X, CHEN H F, WANG Y, WANG Y G, LIU S Y, JIAO C Z,
LU H F, WANG J, YIN C B, JIAO Y L, LU F. Triticum population sequencing provides insights into wheat adaptation. Nature Genetics, 2020, 52(12): 1412-1422.
[24] HAO C Y, JIAO C Z, HOU J, LI T, LIU H X, WANG Y Q, ZHENG J, LIU H, BI Z H, XU F F, ZHAO J, MA L, WANG Y M, MAJEED U, LIU X, APPELS R, MACCAFERRI M, TUBEROSA R, LU H F, ZHANG X Y. Resequencing of 145 landmark cultivars reveals asymmetric sub-genome selection and strong founder genotype effects on wheat breeding in China. Molecular plant, 2020, 13(12): 1733-1751.
[25] LI M X, YEUNG J M Y, CHERNY S S, SHAM P C. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Human Genetics, 2012, 131(5): 747-756.
[26] TEAM R. R: a language and environment for statistical computing. Computer Science, 2014.
[27] YANG J, LEE S H, GODDARD M E, VISSCHER P M. GCTA: a tool for genome-wide complex trait analysis. The American Journal of Human Genetics, 2011, 88(1): 76-82.
[28] LINE R, QAYOUM A. Virulence, aggressiveness, evolution, and distribution of races of(the cause of stripe rust of wheat) in North America 1968-87. US Department of Agriculture Technical Bulletin, 1992, 74, 1788.
[29] CHEN X M. Pathogens which threaten food security: Puccinia striiformis, the wheat stripe rust pathogen. Food e, 2020, 12(2): 239-251.
[30] LIU S J, WANG X T, ZHANG Y Y, JIN Y G, XIA Z H, XIANG M J, HUANG S, QIAO L Y, ZHENG W J, ZENG Q D, WANG Q L, YU R, SINGH R P, BHAVANI S, KANG Z S, HAN D J, WANG C F, WU J H. Enhanced stripe rust resistance obtained by combining Yr30 with a widely dispersed, consistent QTL on chromosome arm 4BL. Theoretical and e Genetics, 2022, 135(1): 351-365.
[31] Van OOIJEN J W. JoinMap4, software for the calculation of genetic linkage maps in experimental populations. Wageningen, The Netherlands, Kyazma BV, 2006.
[32] KOSAMBI D D. The estimation of map distances from recombination values. Annals of Eugenics, 1943, 12(1): 172-175.
[33] VOORRIPS R E. MapChart: Software for the graphical presentation of linkage maps and QTLs. Journal of Heredity, 2002, 93(1): 77-78.
[34] APPELS R, EVERSOLE K, STEIN N, FEUILLET C, KELLER B, ROGERS J, POZNIAK C, CHOULET F, DISTELFELD A, POLAND J, RONEN G, SHARPE A, BARAD O, BARUCH K, KEEBLE-GAGNÈRE G, MASCHER M, BEN-ZVI G, JOSSELIN A, HIMMELBACH A, BALFOURIER F, GUTIERREZ-GONZALEZ J J, HAYDEN M, KOH C, MUEHLBAUER G, PASAM R, PAUX E, RIGAULT P, TIBBITS J, TIWARI V, SPANNAGL M, LANG D, GUNDLACH H, HABERER G, MAYER K, ORMANBEKOVA D, PRADE V M, ŠIMKOVÁ H, WICKER T, SWARBRECK D, RIMBERT H, FELDER M, GUILHOT N, KAITHAKOTTIL G G, KEILWAGEN J, LEROY P, LUX T M, TWARDZIOK S, VENTURINI L, JUHÁSZ A, ABROUK M, FISCHER I, UAUY C, BORRILL P, RAMÍREZ-GONZÁLEZ R, ARNAUD D, CHALABI S, CHALHOUB B, CORY A, DATLA R, DAVEY M, JACOBS J, ROBINSON S J, STEUERNAGEL B, VAN EX F, WULFF B, BENHAMED M, BENDAHMANE A, CONCIA L, LATRASSE D, BARTOŠ J, BELLEC A, BERGÈS H, DOLEŽEL J, FRENKEL Z, GILL B, KOROL A, LETELLIER T, OLSEN O, SINGH K, VALÁRIK M, VAN DER VOSSEN E V D, VAUTRIN S, WEININGS, FAHIMA T, GLIKSON V, RAATS D, ČÍHALÍKOVÁ J, TOEGELOVÁ H, VRÁNA J, SOURDILLE P, DARRIER B, BARABASCHI D, CATTIVELLI L, HERNÁNDEZ P, GÁLVEZ S, BUDAK H, JONES J D G, WITEK K, YU G T, SMALL I, MELONEK J, ZHOU R N, BELOVA T, KANYUKA K, KING R, NILSEN K, WALKOWIAK S, CUTHBERT R, KNOX R, WIEBE K, XIANG D, ROHDE A, GOLDS T, ČÍŽKOVÁ J, AKPıNAR B A, BIYIKLIOGLU S, GAO L L, N'DAIYE A, KUBALAU0301KOVAU0301 M, ŠAFÁŘ J, ALFAMA F, ADAM-BLONDON A, FLORES R, GUERCHE C, LOAEC M, QUESNEVILLE H, CONDIE J, ENS J, MACLACHLAN R, TAN Y F, ALBERTI A, AURY J, BARBE V, COULOUX A, CRUAUD C, LABADIE K, MANGENOT S, WINCKER P, KAUR G, LUO M, SEHGAL S, CHHUNEJA P, GUPTA O, JINDAL S, KAUR P, MALIK P, SHARMA P, YADAV B, SINGH N, KHURANA J, CHAUDHARY C, KHURANA P, KUMAR V, MAHATO A K, MATHUR S, SEVANTHI A, SHARMA N, TOMAR R S S, HOLUŠOVÁ K, PLÍHAL O, CLARK M, HEAVENS D, KETTLEBOROUGH G, WRIGHT J, BALCÁRKOVÁ B, HU Y Q, SALINA E, RAVIN N, SKRYABIN K, BELETSKY A, KADNIKOV V, MARDANOV A, NESTEROV M, RAKITIN A, SERGEEVA E, HANDA H, KANAMORI H, KATAGIRI S, KOBAYASHI F, NASUDA S, TANAKA T, WU J, CATTONARO F, MIN J M, KUGLER K G, PFEIFER M, SANDVE S, XUN X, ZHAN B, BATLEY J, BAYER P, EDWARDS D, HAYASHI S, TULPOVÁ Z, VISENDI P, CUI L C, DU X H, FENG K W, NIE X J, TONG W, WANG L. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science, 2018, 361(6403): eaar7191.
[35] CHEN Y M, SONG W J, XIE X M, WANG Z H, GUAN P F, PENG H R, JIAO Y N, NI Z F, SUN Q X, GUO W L. A collinearity- incorporating homology inference strategy for connecting emerging assemblies in thetribe as a pilot practice in the plant pangenomic era. Molecular plant, 2020, 13(12): 1694-1708.
[36] MA S W, WANG M, WU J H, GUO W L, CHEN Y M, LI G W, WANG Y P, SHI W M, XIA G M, FU D L, KANG Z S, NI F. WheatOmics: A platform combining multiple omics data to accelerate functional genomics studies in wheat. Molecular plant, 2021, 14(12): 1965-1968.
[37] WANG J B, ZHANG Z W. GAPIT version 3: boosting power and accuracy for genomic association and prediction. Genomics, Proteomics & Bioinformatics, 2021, 19(4): 629-640.
[38] YU J M, PRESSOIR G, BRIGGS W H, BI I V, YAMASAKI M, DOEBLEY J F, MCMULLEN M D, GAUT B S, NIELSEN D M, HOLLAND J B, KRESOVICH S, BUCKLER E S. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nature Genetics, 2006, 38(2): 203-208.
[39] LU Y L, ZHANG S H, SHAH T, XIE C X, HAO Z F, LI X H, FARKHARI M, RIBAUT J M, CAO M J, RONG T Z, XU Y B. Joint linkage-linkage disequilibrium mapping is a powerful approach to detecting quantitative trait loci underlying drought tolerance in maize. Proceedings of the National Academy of Sciences of the United States of America, 2010, 107(45): 19585-19590.
[40] ZHANG Y, CUI M, ZHANG J M, ZHANG L, LI C L, KAN X, SUN Q, DENG D X, YIN Z T. Confirmation and fine mapping of a major QTL for aflatoxin resistance in maize using a combination of linkage and association mapping. Toxins, 2016, 8(9): 258.
[41] 常立国, 何坤辉, 刘建超. 多环境下玉米保绿相关性状遗传位点的挖掘. 中国农业科学, 2022, 55(16): 3071-3081.
CHANG L G, HE K H, LIU J C. Mining of genetic locus of maize stay-green related traits under multi-environments. Scientia Agricultura Sinica, 2022, 55(16): 3071-3081. (in Chinese)
[42] ZHANG X X, GUAN Z R, LI Z L, LIU P, MA L L, ZHANG Y C, PAN L, HE S J, ZHANG Y L, LI P, GE F, ZOU C Y, HE Y C, GAO S B, PAN G T, SHEN Y O. A combination of linkage mapping and GWAS brings new elements on the genetic basis of yield-related traits in maize across multiple environments. Theoretical and Applied Genetics,2020, 133(10): 2881-2895.
[43] 刘凯, 邓志英, 张莹, 王芳芳, 刘佟佟, 李青芳, 邵文, 赵宾, 田纪春, 陈建省. 小麦茎秆断裂强度相关性状QTL的连锁和关联分析. 作物学报, 2017, 43(4): 483-495.
LIU K, DENG Z Y, ZHANG Y, WANG F F, LIU T T, LI Q F, SHAO W, ZHAO B, TIAN J C, CHEN J S. Linkage analysis and genome-wide association study of QTLs controlling stem-breaking- strength-related traits in wheat. Acta Agronomica Sinica, 2017, 43(4): 483-495. (in Chinese)
[44] LI W T, ZHU Z W, CHERN M, YIN J J, YANG C, RAN L, CHENG M P, HE M, WANG K, WANG J, ZHOU X G, ZHU X B, CHEN Z X, WANG J C, ZHAO W, MA B T, QIN P, CHEN W L, WANG Y P, LIU J L, WANG W M, WU X J, LI P, WANG J R, ZHU L H, LI S G, CHEN X W. A natural allele of a transcription factor in rice confers broad-spectrum blast resistance. Cell, 2017, 170(1): 114-126.e15.
[45] JEON J E, KIM J G, FISCHER C R, MEHTA N, DUFOUR- SCHROIF C, WEMMER K, MUDGETT M B, SATTELY E. A pathogen-responsive gene cluster for highly modified fatty acids in tomato. Cell, 2020, 180(1): 176-187.e19.
[46] YANG L, HUANG H. Roles of small RNAs in plant disease resistance. Journal of Integrative Plant Biology, 2014, 56(10): 962-970.
[47] WANG J Z, HU M J, WANG J, QI J F, HAN Z F, WANG G X, QI Y J, WANG H W, ZHOU J M, CHAI J J. Reconstitution and structure of a plant NLR resistosome conferring immunity. Science, 2019, 364(6435): eaav5870.
[48] 葸玮, 郝晨阳, 李甜, 刘云川, 焦成智, 王化俊, 张学勇. 基因组时代-麦类基因组学研究现状及趋势. 植物遗传资源学报, 2022, 23(4): 929-942.
XI W, HAO C Y, LI T, LIU Y C, JIAO C Z, WANG H J, ZHANG X Y. The ear genomics: current status and future trend of genomics research triticeae crops. Journal of Plant Genetic Resources, 2022, 23(4): 929-942. (in Chinese)
[49] WALKOWIAK S, GAO L L, MONAT C, HABERER G, KASSA M T, BRINTON J, RAMIREZ-GONZALEZ R H, KOLODZIEJ M C, DELOREAN E, THAMBUGALA D, KLYMIUK V, BYRNS B, GUNDLACH H, BANDI V, SIRI J N, NILSEN K, AQUINO C, HIMMELBACH A, COPETTI D, BAN T, VENTURINI L, BEVAN M, CLAVIJO B, KOO D H, ENS J, WIEBE K, N’DIAYE A, FRITZ A K, GUTWIN C, FIEBIG A, FOSKER C, FU B X, ACCINELLI G G, GARDNER K A, FRADGLEY N, GUTIERREZ-GONZALEZ J, HALSTEAD-NUSSLOCH G, HATAKEYAMA M, KOH C S, DEEK J, COSTAMAGNA A C, FOBERT P, HEAVENS D, KANAMORI H, KAWAURA K, KOBAYASHI F, KRASILEVA K, KUO T, MCKENZIE N, MURATA K, NABEKA Y, PAAPE T, PADMARASU S, PERCIVAL-ALWYN L, KAGALE S, SCHOLZ U, JUN S S, JULIANA P, SINGH R, SHIMIZU-INATSUGI R, SWARBRECK D, COCKRAM J, BUDAK H, TAMESHIGE T, TANAKA T, TSUJI H, WRIGHT J, WU J Z, STEUERNAGEL B, SMALL I, CLOUTIER S, KEEBLE-GAGNÈRE G, MUEHLBAUER G, TIBBETS J, NASUDA S, MELONEK J, HUCL P J, SHARPE A G, CLARK M, LEGG E, BHARTI A, LANGRIDGE P, HALL A, UAUY C, MASCHER M, KRATTINGER S G, HANDA H, SHIMIZU K K, DISTELFELD A, CHALMERS K, KELLER B, MAYER K F X, POLAND J, STEIN N, MCCARTNEY C A, SPANNAGL M, WICKER T, POZNIAK C J. Multiple wheat genomes reveal global variation in modern breeding. Nature, 2020, 588(7837): 277-283.
[50] SHI X L, CUI F, HAN X Y, HE Y L, ZHAO L, ZHANG N, ZHANG H, ZHU H D, LIU Z X, MA B, ZHENG S S, ZHANG W, LIU J J, FAN X L, SI Y Q, TIAN S Q, NIU J Q, WU H L, LIU X M, CHEN Z, MENG D Y, WANG X Y, SONG L Q, SUN L J, HAN J, ZHAO H, JI J, WANG Z G, HE X Y, LI R L, CHI X B, LIANG C Z, NIU B F, XIAO J, LI J M, LING H Q. Comparative genomic and transcriptomic analyses uncover the molecular basis of high nitrogen-use efficiency in the wheat cultivar Kenong 9204. Molecular Plant, 2022, 15(9): 1440-1456.
[51] CHENG H, LIU J, WEN J, NIE X J, XU L H, CHEN N B, LI Z X, WANG Q L, ZHENG Z Q, LI M, CUI L C, LIU Z H, BIAN J X, WANG Z H, XU S B, YANG Q, APPELS R, HAN D J, SONG W N, SUN Q X, JIANG Y. Frequent intra- and inter-species introgression shapes the landscape of genetic variation in bread wheat. e Biology, 2019, 20(1): 136.
[52] LI A L, HAO C Y, WANG Z Y, GENG S F, JIA M L, WANG F, HAN X, KONG X C, YIN L J, TAO S, DENG Z Y, LIAO R Y, SUN G L, WANG K, YE X G, JIAO C Z, LU H F, ZHOU Y, LIU D C, FU X D, ZHANG X Y, MAO L. Wheat breeding history reveals synergistic selection of pleiotropic genomic sites for plant architecture and grain yield. Molecular Plant, 2022, 15(3): 504-519.
[53] RAMÍREZ-GONZÁLEZ R H, BORRILL P, LANG D, HARRINGTON S A, BRINTON J, VENTURINI L, DAVEY M, JACOBS J, VAN EX F, PASHA A, KHEDIKAR Y, ROBINSON S J, CORY A T, FLORIO T, CONCIA L, JUERY C, SCHOONBEEK H, STEUERNAGEL B, XIANG D, RIDOUT C J, CHALHOUB B, MAYER K X, BENHAMED M, LATRASSE D, BENDAHMANE A, CONSORTIUM I W G S, WULFF B H, APPELS R, TIWARI V, DATLA R, CHOULET F, POZNIAK C J, PROVART N J, SHARPE A G, PAUX E, SPANNAGL M, BRÄUTIGAM A, UAUY C. The transcriptional landscape of polyploid wheat. e, 2018, 361(6403): eaar6089.
[54] KELLER B, WICKER T, KRATTINGER S G. Advances in wheat and pathogen genomics: Implications for disease control. Annual Review of Phytopathology, 2018, 56: 67-87.
[55] YANG X, ZHONG S B, ZHANG Q J, REN Y, SUN C W, CHEN F. A loss-of-function of the dirigent gene TaDIR-B1 improves resistance to Fusarium crown rot in wheat. Plant e Journal, 2021, 19(5): 866-868.
[56] CHEN X M, KANG Z S. Stripe Rust. Dordrecht: Springer Netherlands Press, 2017: 723.
Identification of Adult Plant Stripe Rust Resistance Candidate Genes ofby Gene Association and Transciptome Analysis in Wheat (L.)
1College of Agronomy, Northwest A&F University/State Key Laboratory of Crop Stress Biology for Arid Areas, Yangling 712100, Shaanxi;2College of Plant Protection, Northwest A&F University/State Key Laboratory of Crop Stress Biology for Arid Areas, Yangling 712100, Shaanxi
【Objective】Stripe rust, caused byf. sp.(), significantly reduced wheat production worldwide. Identification of stripe rust resistance genes is the foundation of improving wheat resistance breeding and revealing its genetic mechanism.【Method】A multi-omics approach combined with genome-wide association study (GWAS) was used for dissecting adult plant stripe rust resistance for wheat advanced breeding lines collected from International Maize and Wheat Improvement Center (CIMMYT) and International Centre for Agricultural Research in the Dry Areas (ICARDA) bread-wheat breeding programs. In the present study, a diversity panel of 411 wheat lines from CIMMYT and ICARDA was used for genome-wide association study and a major locus on chromosome arm 2BL was identified. In order to verify the stability of the locus, the resistant line Z501 with the resistance allele of the locus was crossed by the susceptible line Jinmai 79, and the locus tentatively namedwas successfully confirmed using linkage mapping based on F2:3genetic population of Jinmai 79×Z501. Then we performed candidate gene analysis based on gene annotation, comparative genome, transcriptome and gene-based association analysis. 【Result】Combining GWAS and linkage mapping results, thewas located in the physical interval of 0.26 Mb (575.706-576.587 Mb) on chromosome 2B. According to the annotation information of Chinese Spring reference genome IWGSC v1.1, there were six high confidence genes of 12 genes in this region. Using online website, the target interval in the Chinese spring reference genome was compared with other published different ploidy wheat genomes. The six high-confidence genes within this interval can basically be found homologous in other wheat lines, and the genes arranged in the same order, indicating that the interval may not have large fragment insertions, deletions and inversions. The above results showed that we can perform candidate gene prediction analysis based on the reference genome information. After analysis of their transcriptomic data between the resistant parent Z501 and susceptible parent Jimai 79, only three genes,,andshowed variable expression levels and were induced by stripe rust infection. Further, they encode GATA transcription factor, SH3 domain-containing protein 2 and zinc finger protein, respectively. Gene-based association analysis revealed that there was a significant SNP (G1369A) inthat was associated with stripe rust responses. Although this SNP (G1369A) did not cause amino acid coding changes (both TCG and TCA encode serine), it may be associated with alternative splicing. Moreover, it showed significant differences of the stripe rust responses between the different haplotypes (G1369A). Further analysis revealed two other variants G1377A and G1431A, that caused amino acid changes, i. e. valine (GTT) to isoleucine (ATT) and valine (GTG) to methionine (ATG), respectively. However, the two SNPs were rare variants as they accounting for only 0.87% of the 455 re-sequencing wheat accessions and they were not tested for significance. In summary,was preliminarily considered as an important candidate gene of. In addition, the corresponding AQP markers were developed based on the SNPs among thecandidate regions, which can be used to marker-assisted selection in molecular breeding application of wheat stripe rust resistance.【Conclusion】A candidate causal geneassociated with stripe rust resistance was successfully identified on wheat chromosome 2B using an integrated method of multi-omics and association analysis, which laid a solid foundation for further gene cloning and functional verification.
; stripe rust resistance;; integrative analysis of multiple omics; SH3P2
10.3864/j.issn.0578-1752.2023.08.001
2022-12-08;
2023-01-19
国家重点研发计划(2021YFD1200600)、青海省重点研发与转化计划(2022-NK-125)
张旭,E-mail:zhxu@nwafu.edu.cn。韩金妤,E-mail:1021979347@qq.com。张旭和韩金妤为同等贡献作者。通信作者韩德俊,E-mail:handj@nwsuaf.edu.cn。通信作者吴建辉,E-mail:wujianhui@nwsuaf.edu.cn
(责任编辑 李莉)
