Amplification and Bioinformatics Analysis of vscN Gene from Vibrio alginolyticus
2023-06-27LinlinYINWenLIHaiyunFENGWeijieZHANGJunlinWANGHuanyingPANGNaWANG
Linlin YIN, Wen LI, Haiyun FENG, Weijie ZHANG, Junlin WANG, Huanying PANG*, Na WANG
1. College of Fisheries, Guangdong Ocean University, Zhanjiang 524025, China; 2. Guangdong Provincial Key Laboratory of Aquatic Animal Disease Control and Healthy Culture / Key Laboratory of Control for Diseases of Aquatic Economic Animals of Guangdong Higher Education Institutes, Zhanjiang 524025, China; 3. Chinese Academy of Inspection and Quarantine, Beijing 100176, China
Abstract [Objectives] The paper was to clone and analyze bioinformatics of vscN gene from Vibrio alginolyticus. [Methods] A pair of specific primers was designed based on the vscN gene sequence of V. alginolyticus HY9901. The full length of vscN gene was amplified by PCR and bioinformatics analysis was performed. [Results] The vscN gene was 1 323 bp in total, encoding 440 amino acids, with the theoretical molecular weight of 47.86 kD and the theoretical pI value of 5.89. The online prediction showed that there was no signal peptide and no transmembrane region in VscN. The amino acid sequence had 10 N-myristoylation sites, 8 phosphorylation sites (2 protein kinase C phosphorylation sites, 6 casein kinase II phosphorylation sites), 1 amidation site, 11 microbody C-terminal target signal sites, 1 ATP/GTP binding site motif A (P ring), and 1 ATPase α and β subunit specific site. Homology analysis showed that the VscN protein of V. alginolyticus had high homology with that of V. antiquarius, with a similarity of 95.14%. Phylogenetic tree analysis showed that the VscN of V. alginolyticus was clustered into the same subgroup as that of V. diabolicus and V. antiquarius. Functional domain analysis of VscN protein showed that it had Pfam and AAA domains, and involved in the regulation of bacterial virulence. The three-dimensional structure model of VscN simulated by SWISS-MODEL software was similar to the structure of flagellate-specific ATPase FliH-FliI complex. [Conclusions] The results lay a foundation for further study on the regulatory mechanism of VscN protein on bacterial virulence.
Key words Vibrio alginolyticus, T3SS, VscN, Bioinformatics analysis
1 Introduction
Vibrioalginolyticusis a kind of halophilic, thermophilic, facultative anaerobic gram-negative brevibacterium, common in marine environment and a variety of culture environments. WhenV.alginolyticusinfects mariculture animals such as fish, shrimp and shellfish, it is easy to cause large-scale death of aquaculture animals, resulting in huge economic losses. In addition, the bacterium can also cause food poisoning, otitis media, headache and sepsis in humans[1]. In recent years, the prevention and control ofV.alginolyticusin China mainly relies on antibiotics, but the long-term use of antibiotics will lead to drug residues, bacterial resistance,etc.It will not only increase the difficulty in the prevention and control of various epidemics in the breeding process, but also affect the quality and safety of aquatic products and ecological environment safety, ultimately threatening human health[1-4]. Therefore, it is necessary to explore the pathogenic genes and action mechanism ofV.alginolyticusfor the prevention and control of aquatic animals.
Type III secretion system (T3SS) is found in gram-negative bacteria, such asYersinia[5],Salmonella[6-7]andPseudomonas[8]. WhenV.alginolyticusinfects the host, T3SS can secrete virulence proteins outside the cells or on the surface of host cells, or even directly inject virulence proteins into host cells, resulting in cell death[9-10].
ATPase of T3SS exerts ATPase activity in the form of monomer[11-12], and VscN protein, as the ATPase inVibriotype III secretory system[13], is closely related to the virulence ofVibrio[14]. At the same time, VscN protein can also mediate dissociation of protein from its molecular chaperone conjugate[15]. Zhuang Qiutingetal.[11]suggested that VscN protein inV.splendidushad ATPase activity and was closely related to the virulence protein secreted byV.splendidus. Bill Blaylocketal.[16]put forward that EscN protein in enterohemorrhagicEscherichiacolialso had ATPase activity and provided an important energy source in the process of secreting effectors protein by T3SS. However, until now, there has been no report on thevscNgene ofV.alginolyticus. In this study, we cloned thevscNgene, the DNA-binding response regulatory factor ofV.alginolyticus, and analyzed its sequence characteristics and subunit structure, which would lay a foundation for further study on the regulatory mechanism of VscN protein on bacterial virulence.
2 Materials and methods
2.1 MaterialsV.alginolyticusHY9901 was isolated from infectedLutjanussanguineusin Zhanjiang sea area of Guangdong Province, and preserved[17]. The clone vector pMD18-T was purchased from Takara.
ExTaqDNA polymerase was purchased from Takara. Bacterial genome DNA extraction kit and DNA glue recovery kit were purchased from Tiangen Biotech Co., Ltd., and other reagents were imported or domestic analytical pure. PCR primer synthesis and sequencing were completed by Sangon Biotech (Shanghai) Co., Ltd. The antibiotic ampicillin (Amp+) was used at a concentration of 100 μg/mL.
The instruments used in the test included electrophoresis meter and PCR meter (Bio-rad Laboratories), ultra pure water instrument (Beijing Rightleder Water Treatment Equipment Co., Ltd.), refrigerated high-speed centrifugator (Eppendorf), HVE-50 series autoclave (Hirayama), gel imaging equipment (Priotein Simple), ultraviolet spectrophotometer (Shimadzu), ultra low temperature freezer (Theromo Fisher Scientific).
2.2 Methods
2.2.1Extraction of genome fromV.alginolyticusHY9901.V.alginolyticuswas taken from an ultra low temperature freezer and streaked on TSA plate. Single clones were selected and inoculated in TSB medium at a ratio of 1:100. After cultured in a shaker at 28 ℃, 180 r/min for more than 14 h, a small amount of bacterial liquid was transferred into centrifuge tubes. The tube were centrifuged at 10 000 r/min for 2-3 min. The supernatant was discarded andV.alginolyticuswas collected. Total DNA ofV.alginolyticuswas extracted according to the bacterial genome extraction kit, and refrigerated at -20 ℃ for later use.
2.2.2Cloning ofvscNgene. A pair of primers was designed according to thevscNgene sequence ofV.alginolyticusin Genbank. Forward primer: 5’-ATGGAACAACAGGCAATCAAT-3’; reverse primer: 5’-GTGTCTTGGTTGAAACTTATATCC-3’. PCR was performed in the following procedures: pre-denaturating at 95 ℃ for 5 min; denaturating at 95 ℃ for 30 s, annealing at 60 ℃ for 30 s, extension at 68 ℃ for 30 s, 30 cycles; extension at 72 ℃ for 10 min. PCR products were detected by electrophoresis on 1% agarose gel and purified by gel cutting kit.
2.2.3Connection and sequencing of target fragment and vector. ThevscNand pMD 18-T vectors were connected overnight at 4 ℃, and the connected products were transferred intoE.coliDH5α competent cells for incubation at 37 ℃. After colony PCR assay, the positive clones were sequenced.
2.2.4Website for bioinformatics analysis[18]. ExPASy Proteomics Server was used to predict the amino acid sequence, theoretical isoelectric point (pI) and molecular mass of target protein online. SignalP 4.1 Server was applied to predict whether the target protein contained signal peptide. GeneDoC software was used to align the homology of amino acid sequences of target proteins. Clastal 2.0 and MEGA 6.0 software were adopted to construct the phylogenetic tree. TMHMM Server 2.0 was employed to predict whether the target protein contained transmembrane structure. SoftBerry-Psite was used to analyze functional sites in amino acid sequences of proteins. NCBI conserved domain database CDD was adopted to predict the domains of target proteins. SWISS-MODEL was employed to predict and construct the tertiary structure of proteins online.
3 Results and analysis
3.1 Full-length cloning ofvscNgeneThevscNgene was amplified by PCR assay. The amplified products were analyzed by agarose gel electrophoresis, and specific bands were amplified (Fig.1). Sequencing of the amplified product and the cloned vector pMD18-T showed that thevscNgene contained an open reading frame of 1 323 bp, encoding 441 amino acids. The gene of the amplified product was submitted to GenBank (vscNaccession number: MN328350.1).
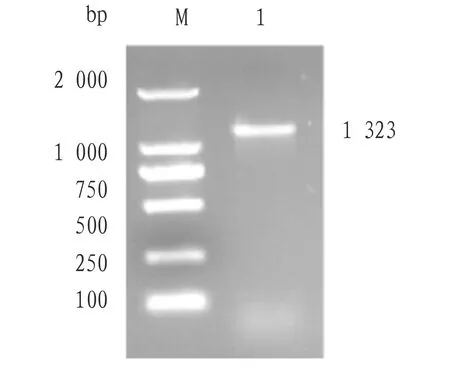
Fig.1 Cloning of vscN gene
3.2 Physicochemical properties of VscNThe physicochemical properties of VscN protein ofV.alginolyticuswas analyzed by ExPASy software. The results showed that the total number of atoms was 6 766 and the molecular structure formula was C2099H3408N602O640S17. The theoretical molecular weight was 47.86 kD and the theoretical pI was 5.89. The instability coefficient was 37.02 (threshold value 40), so the protein was relatively stable. The fat coefficient was 96.64, the total mean hydrophilicity was -0.145, and the total hydrophobicity of the protein was low. The total number of acid amino acid residues (Asp+Glu) was 56, and that of basic amino acid residues (Arg+Lys) was 56, and Leu was at the N terminus. The half-lifeinvivowas 3 min in yeast and 2 min inE.coli, and the half-lifeinvitrowas 5.5 h in mammalian reticular cells.
3.3 VscN sequence analysisThe N-terminal signal peptide structure of the amino acid sequence ofvscNgene was predicted by SignalP 4.0 Server program, and no signal peptide was found in the gene. Prediction made by TMHMM Server 2.0 program showed that the protein had no transmembrane region. Prediction by SoftBerry-Psite showed that the amino acid sequence contained 10 N-myristoylation sites, 8 phosphorylation sites (2 protein kinase C phosphorylation sites, 6 casein kinase II phosphorylation sites), 1 amidation site, 11 microbody C-terminal target signal sites, 1 ATP/GTP binding site motif A (P ring), and 1 ATPase α and β subunit specific site (Fig.2).

Note: : Casein kinase II phosphorylation site; : Microbody C-terminal target signal site; : Protein kinase C phosphorylation site; : ATP/GTP binding site motif A (P ring); : ATPase α and β subunit specific site; : N-myristoylation site; : Amidation site.Fig.2 The nucleotide of vscN gene and its encoded amino acid sequence
3.4 Homology and evolutionary analysisHomology analysis was performed using DNAMAN software. The VscN protein sequences ofV.harveyi,V.paracholerae,V.diabolicus,V.antiquarius,V.jasicida,V.cholerae,V.mimicusand otherVibriostrains were aligned with that ofV.alginolyticus. The results showed that the VscN ofV.alginolyticushad high homology with that ofV.antiquarius, with a similarity of 95.14% (Fig.3). The phylogenetic tree of VscN amino acid sequences of the derivedV.antiquariusand otherVibriostrains was constructed by Neighbor-joining method of MEGA 5.0, and the results showed thatV.antiquariuswas clustered into the same subgroup withV.diabolicusandV.antiquarius(Fig.4).
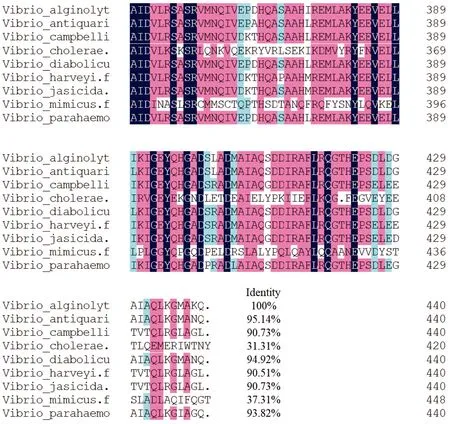
Fig.3 Homology comparison of amino acid sequences derived from vscN gene
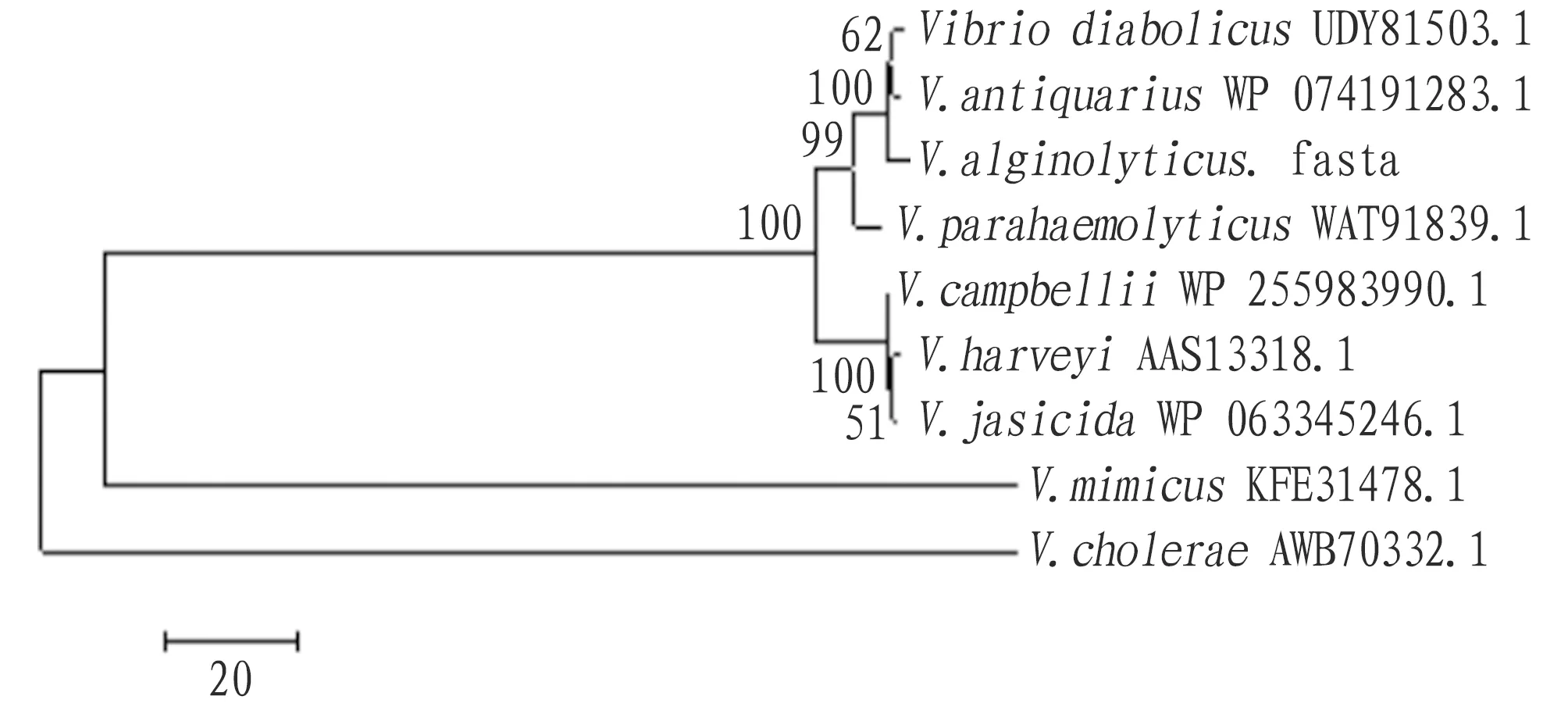
Fig.4 Phylogenetic tree of VscN amino acid sequence based on NJ method
3.5 Prediction of functional domain and secondary structure
The functional domains of amino acid structure were analyzed using the SMART website program, and the prediction results showed that VscN protein had two functional domains, namely Pfam and AAA(Fig.5). At the N-terminus of VscN protein, transmembrane ATPase is a membrane binding enzyme complex/ion transporter that uses ATP hydrolysis to drive transmembrane transport of protons. Some transmembrane ATPase also work in reverse, and drive ATP synthesis by using the energy of proton gradient and the transmembrane ion concentration difference when ions pass through the ATPase proton channel. At the C-terminus of VscN protein, the AAA+ superfamily of ATPase is present in all organisms and is involved in a variety of cellular processes, including membrane fusion, proteolysis, and DNA replication. The prediction results of secondary structure of VscN protein showed that the α-helix, random coil, extended strand and β-sheet accounted for 45.00%, 33.18%, 14.09% and 7.73%, respectively (Fig.6).
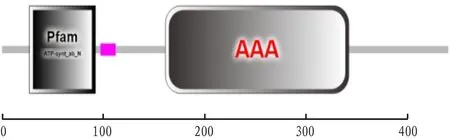
Fig.5 Functional domain of VscN protein
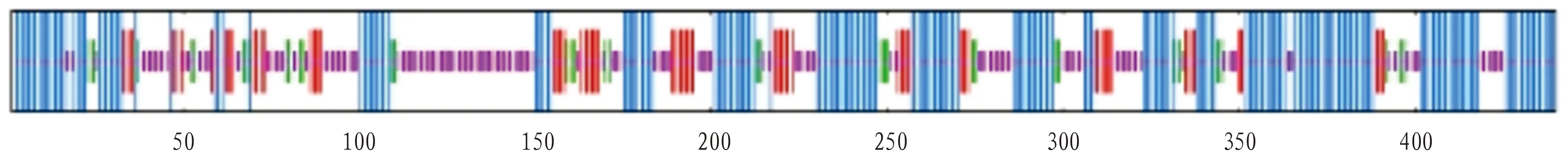
Fig.6 Secondary structure prediction of VscN protein
3.6 Tertiary structure of VscN proteinSwiss-model software was used for homology modeling of VscN amino acid sequences, and corresponding tertiary structure prediction model was obtained (Fig.7). It was close to the model of flagella-specific FliH-FliI complex structure (SMTL ID: 5b0o.1), with a similarity of 42.43%.
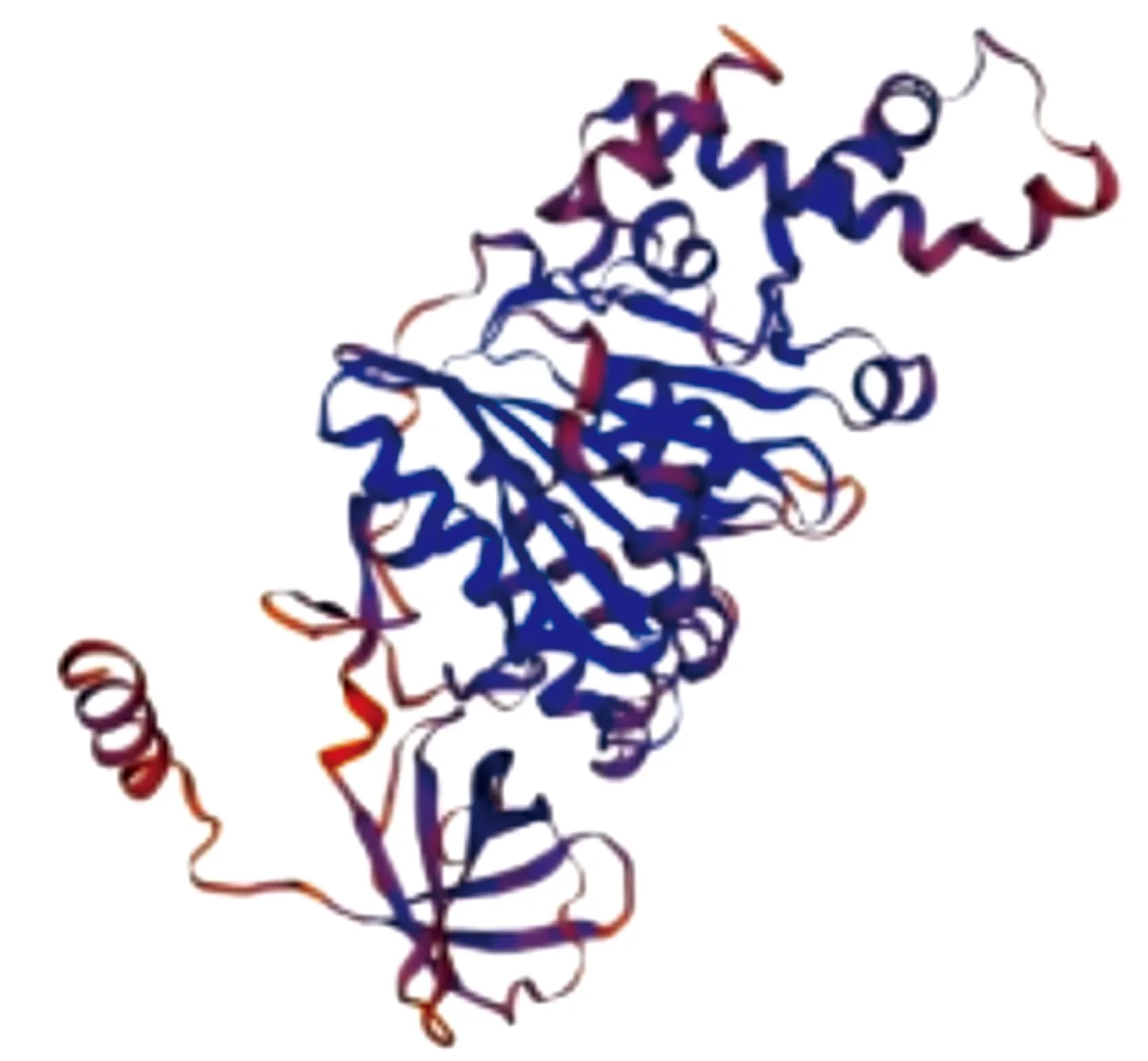
Fig.7 Tertiary structure prediction of VscN protein
4 Discussion
At present, bioinformatics analysis is an important method to predict and analyze the functional structure of proteins, and can accurately predict the physicochemical properties and advanced structure of proteins[19]. In this study, thevscNgene ofV.alginolyticuswas cloned successfully and bioinformatics analysis was performed. ThevscNgene was 1 323 bp in total, encoding 440 amino acids, with the theoretical molecular weight of 47.86 kD and the theoretical pI value of 5.89. The online prediction showed that thevscNgene had no signal peptide, 10 N-myristoylation sites, 8 phosphorylation sites (2 protein kinase C phosphorylation sites, 6 casein kinase II phosphorylation sites), 1 amidation site, 11 microbody C-terminal target signal sites, 1 ATP/GTP binding site motif A (P ring), and 1 ATPase α and β subunit specific site. In this study, the tertiary structure of VscN protein was simulated, which was similar to the structure of flagellate-specific ATPase FliH-FliI complex. Homology analysis illustrated thatV.alginolyticushad relatively close genetic relationship withV.diabolicusandV.antiquarius. Functional domain analysis of VscN protein showed that it had Pfam and AAA domains, which involved in the regulation of bacterial virulence.
Type III secretory system is widely found in pathogenic bacteria and plays an important role in the pathogenic process of bacteria to host[20]. When bacteria infect the host, they will inject virulence factors contained in effector proteins into host cells together, and mediate the activation of virulence in host cells through effector proteins[21]. In the acicular complex assembled by T3SS, highly conserved ATPase is an important core element and is located at the base of acicular complex[12,21]. ATPase is thought to be involved in the output of effector proteins, including the recognition, transport and secretion of effector proteins, thus indirectly controlling host cell lysis and interfering with host cell function.
Bioinformatics analysis predicted that VscN protein had no transmembrane region, and it may catalyze the decomposition of ATP and provide energy for T3SS through its own ATPase activity, and play the role of affecting the transcription and expression of bacteria-related virulence genes, regulating the adaptability of bacteria to the environment, and resisting the adverse environment for its growth and reproduction, thus resisting the phagocytosis and killing of host immune system. WhenVibriostrains infect other cells, ATPase in T3SS can hydrolyze ATPase intracellularly, providing energy for the secretion process of effector proteins[11-12]. It will further affect vesicle transport, immune response and other physiological activities of host cells, so as to facilitate more efficient infection of cells. At present, there are rare reports on thevscNgene ofV.alginolyticus, whereas this gene is related to virulence. Hence, further study of this gene will play a positive role in the prevention and control of vibriosis and improving the current breeding environment of aquatic economic animals.
杂志排行

Asian Agricultural Research的其它文章
- Construction of Industrialization Development Model for "1+4+X" Community-Based Elderly Care in Gannan Old Revolutionary Base Area in the Context of Population Aging
- Preparation of Biomanganese Oxide-biochar Composite and Its Remediation of Arsenic Contamination
- Flower Characteristics and Resource Evaluation of Fruit Mulberry