基于XGBoost的启动子及其类型识别的两层预测器
2023-06-25胡仔豪



摘 要:启动子的分类已成为一个有趣的问题,并引起了生物信息学领域许多研究人员的关注。为解决这一问题,进行了多种研究,但其性能结果仍需进一步改进。为此,基于机器学习和深度学习算法,引入了一种智能计算模型,即iPSI(2L)-XGBoost,用于区分启动子及其强弱。所提出的计算模型iPSI(2L)-XGBoost能够在两层中分别达到86.79%和78.64%的交叉验证精度,就所有评估指标而言,拟议的iPSI(2L)-XGBoost模型比其他模型获得了有效的成功率。因此,iPSI(2L)-XGBoost模型将为启动子鉴定的学术研究提供一个有用的工具。
关键词:启动子;启动子识别;卷积神经网络;多特征融合;XGBoost
中图分类号:TP39;TP18 文献标识码:A 文章编号:2096-4706(2023)07-0078-04
Abstract: The classification of promoters has become an interesting issue and has attracted the attention of many researchers in the field of bioinformatics. To solve this problem, various studies have been conducted, but their performance results still need to be further improved. Therefore, based on machine learning and deep learning algorithms, an intelligent computing model, iPSI(2L)-XGBoost, is introduced to distinguish promoters and their strengths. The proposed computing model iPSI(2L)-XGBoost can achieve cross validation accuracy of 86.79% and 78.64% in two layers, respectively. For all evaluation indicators, the proposed iPSI(2L)-XGBoost model achieves an effective success rate compared to other models. Therefore, the iPSI(2L)-XGBoost model will provide a useful tool for academic research on promoter identification.
Keywords: promoter; promoter recognition; Convolutional Neural Networks; Multi-feature fusion; XGBoost
0 引 言
DNA中的启动子是基因的重要组成部分,它调节特定细胞中特定基因的转录。与真核生物基因表达调控相比,原核生物基因表达调节是简单的。在前一种情况下,两个调节过程,即转录和翻译同时发生,而在后一种情况中,基因表达调节是一种更复杂的现象,因为最初的DNA合成发生在转录和翻译之后。更重要的是,在原核生物中,大多数基因都由一个操纵子控制,操纵子将大多数基因作为一个表达簇进行调控和转录。染色体上的特定区域决定了特定转录物的命运,是否或如何启动转录。这些序列被称为启动子,对基因表达调控和控制特定途径至关重要,其位于转录起始位点(TSS)附近。RNA聚合酶(RNAP)和各种称为“σ-因子”的蛋白质的组合可以通过诱导RNA全酶来确定TSS[1],因此,σ-因子可以用于识别原核启动子区域。根据σ-因子(例如,大肠杆菌中的σ70、σ54、σ38、σ32、σ28和σ24)[2]及其转录激活和表达水平的强度不同,启动子区域结构谱中的模式可分为几类[3,4]。原核生物中只有一种RNA聚合酶(RNAP)可以转录其基因,但有多种σ因子[5]。启动子也可分为强启动子和弱启动子,强启动子可提高转录频率和外源基因表达水平。据我们所知,iPSW(2L)-PseKNC[6]是识别启动子强弱的第一个预测器,DNA序列是由伪K核苷酸组成(PseKNC)融合核苷酸密度和这些物理化学性质形成的,使用支持向量机进行预测。最近,为了确定启动子及其强弱,已经做出了许多努力。Le等人使用语言模型提取DNA序列的特征,然后将其输入卷积神经网络(CNN)进行分类,发现FastText在这个问题上比Glove或Word2Vector取得了更好的性能[7]。Tayara等人提出了iPSW(PseDNC-DL)模型,该模型结合了从卷积层和PseDNC中提取的特征[8]。这里,我们使用新构建的特征提取方法,即One-hot、PCA-PseKNC和PseKNC三种编码方法融合后再利用XGBoost分类算法进行预测分类,得到一个新的两层预测器iPSI(2L)-XGBoost,以提高识别启动子及其强度类型的预测能力。
1 材料与方法
1.1 基准数据集
收集高质量数据集是解决生物信息学问题的最重要步骤之一,现用于预测启动子强弱类型的数据集均是基于Xiao[6]等人提出的数据集。为了客观评估我们的模型与其他现有模型之间的性能差异,我们也使用了iPSW(2L)-PseKNC[6]模型構建的基准数据集,并将其命名为数据集Ⅰ。在这个数据集中,他们从RegulonDB[9]收集了所有实验证实的启动子序列,这是基因表达调控网络的巨大数据库。这些序列根据其转录激活和表达水平分为两类:强启动子和弱启动子。他们还通过考虑内含子、外显子和基因间序列(不包括阳性序列)来提取非启动子序列。之后,CD-HIT[10]也被用来排除相似度超过85%的成对序列。数据集Ⅰ包括3 382个启动子样本和3 382个非启动子样本。在3 382个启动子样本中,有1 591个强启动子样本和1 792个弱启动子样本用于构建二级分类。
1.2 特征提取
对于数据集中给定的DNA序列样本,可以表示为:
DNA由四种不同类型的核苷酸组成:A、C、G和T,其中A是腺嘌呤,C是胞嘧啶,G是鸟嘌呤,T是胸腺嘧啶。在One hot编码中,A编码为(1,0,0,0),C编码为(0,1,0,0),G编码为(0,0,1,0),T编码为(0,0,0,1)。伪K元组核苷酸合成(PseKNC)是一种结合核苷酸的物理化学性质和频率密度的编码方法。每个核苷酸都有不同的物理化学性质,例如化学结构、内部结合特征以及根据互补碱基配对原理,都可以分成不同的类别。
根据不同的物理化学性质,四个核苷酸可以用不同的编码表示,在PseKNC特征编码中,第i个核苷酸可以编码为(xi, yi, zi, pi),其中第i个核苷酸分别为A、C、G、T时其理化性质编码为(1,1,1),(0,1,0),(1,0,0)和(0,0,1)。
在本研究中,我们构建了一种新的编码方法,称为PCA-PseKNC。PCA-PseKNC的构建原理与PseKNC相同,只是将one-hot编码改为核苷酸二联体编码。
我们提出了一种基于卷积神经网络、PCA-PseKNC和PseKNC的特征融合编码方法。图1给出了如何获得序列特征的流程图。利用卷积神经网络从One-hot和PseKNC融合后的特征编码中提取重要特征。其主要思想是通过使用卷积和池化层来实现数据特征的自学习,以及降低特征的維数。
基于网格搜索算法,选择各层CNN模型的超参数配置。网格搜索算法是一种超参数优化算法,通过它可以从超参数空间的手动定义子集中选择最佳的超参数组合。调谐的超参数是卷积和稠密层后的丢失概率、卷积层的数量、滤波器的数量、滤波器的大小、最大池化层。表1给出了第一层和第二层启动子和启动子强度识别中使用的CNN模型的详细配置。
式(4)表示卷积运算“Conv1D”其中D表示DNA序列的输入,f表示核的指数,j表示输出位置的指数。卷积的每个滤波器W f由一个权重矩阵S×N表示,其中S是滤波器尺寸,N是输入通道的数量。密集层如式(5)所示,其中xi是1×d维特征向量,wi是前一层xi的权重,wd+1是一个加性偏差项。式(6)包含密集层的脱落操作,其中mi是从概率为α的贝努利分布中取样的。为了避免网络过拟合,应用了Dropout正则化,ReLU是一个激活函数,它表示校正的线性函数,并在数学上用式(7)表示。式(8)中表示的sigmoid,用于预测所提供的序列是启动子还是非启动子。该层的输出通过一个sigmoid函数缩放到[0,1],其中x是这些函数的输入。
在这项研究中,使用CNN模型去提取一个新的特征,然后再将这组特征与PCA_PseKNC特征融合从而获得一个新的特征编码,最后将输出特征编码展平以获得最终的特征编码。对每个序列形成的二维矩阵进行扩展,得到最终的880维特征编码。样本序列D可以表示为:
1.3 预测模型
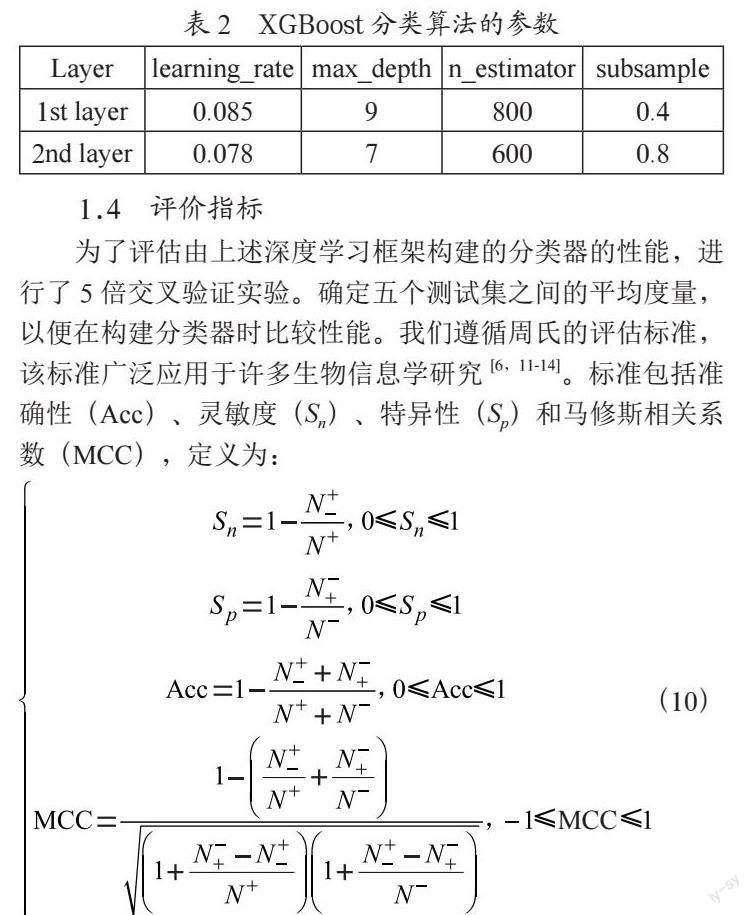
本研究使用XGBoost作为构建预测器的分类算法,故将预测器命名为iPSI(2L)-XGBoost,该计算模型中的一些参数如表2所示。在预测过程中,使用机器学习和深度学习算法,以区分第一层中的启动子或非启动子,而第二层对启动子进行了强启动子或弱启动子的预测。
1.4 评价指标
为了评估由上述深度学习框架构建的分类器的性能,进行了5倍交叉验证实验。确定五个测试集之间的平均度量,以便在构建分类器时比较性能。我们遵循周氏的评估标准,该标准广泛应用于许多生物信息学研究[6,11-14]。标准包括准确性(Acc)、灵敏度(Sn)、特异性(Sp)和马修斯相关系数(MCC),定义为:
其中N +是所调查的阳性样本或启动子的总数;N -是调查的阴性样品或非启动子的总数; 是错误地预测为启动子的非启动子的数量以及 是错误地预测为非启动子的启动子样本的数量。同样,我们还使用接收器工作特性(ROC)曲线下的面积AUC[15]作为性能评估的附加指标。AUC是一个介于0到1之间的概率值,其中较大的AUC表示更好的预测性能。
2 结果和讨论
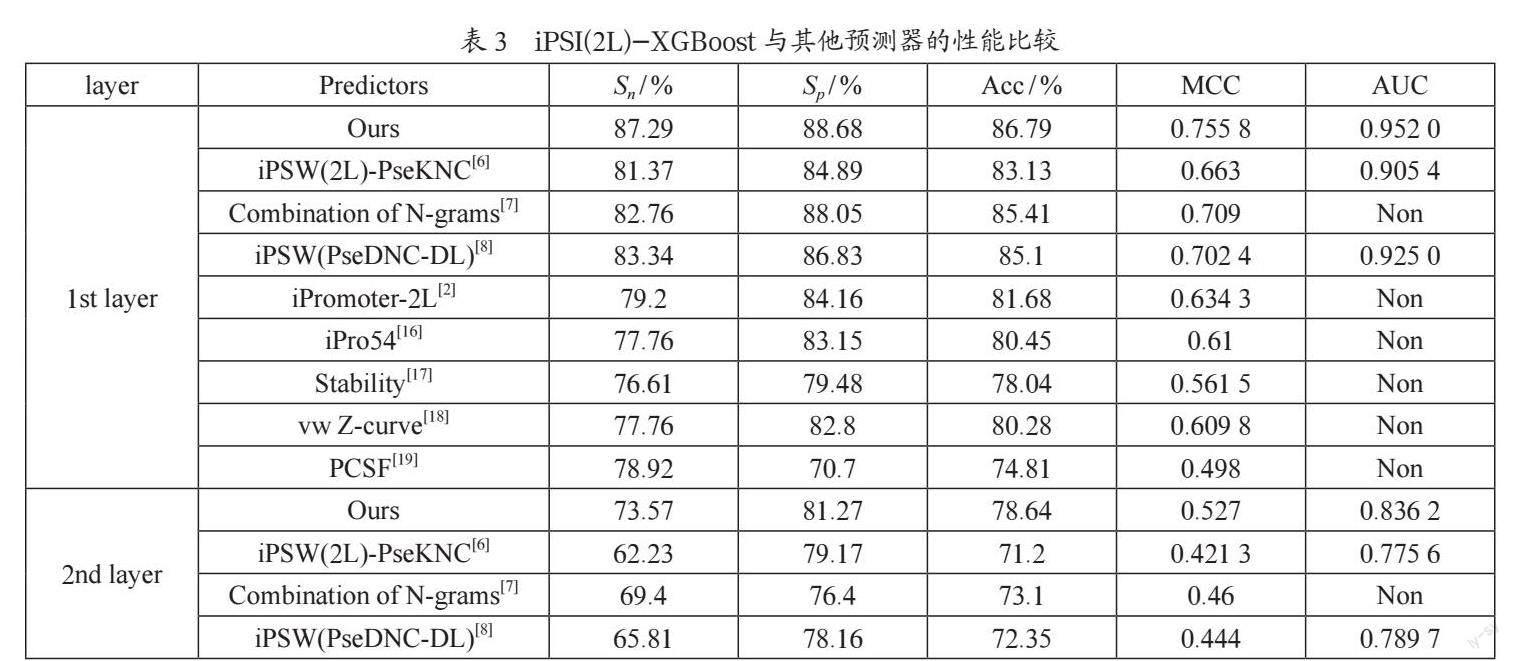
K折叠交叉验证应用非常广泛,非常适合于大型数据集,因为它可以减少计算时间。用于预测启动子强弱类型的现有预测器均基于5倍交叉验证进行了性能检查。为了与现有模型进行比较,我们也使用了5倍交叉验证方法,并用五个指标评估了预测器的性能:灵敏度(Sn)、特异性(Sp)、准确度(Acc)、马修相关系数(MCC)和ROC曲线下面积(AUC)。就鉴定是否为启动子而言,iPSI(2L)-XGBoost获得的Sn、Sp、Acc、MCC和AUC分别为87.29%、88.68%、86.79%、0.755 8和0.952;对于启动子的预测强度,Sn、Sp、Acc、MCC和AUC分别为73.57%、81.27%、78.64%、0.547和0.836 2。
为了进一步证明iPSI(2L)-XGBoost预测器的功效,将其与现有预测器进行了比较,表3显示了iPSI(2L)-XGBoost和其他预测器之间的性能比较,本研究中提出的预测器优于其他预测器。在同一数据集上,iPSI(2L)-XGBoost在两个任务中都优于iPSW(2L)-PseKNC。在启动子识别任务中,与其他的最佳预测器Combination of N-grams[7]相比,iPSW(2L)-XGBoost的Acc提高了1.38%,MCC提高了4.68%。在启动子强度识别中,iPSI(2L)-XGBoost分别使Acc和MCC提高了5.54%和6.7%。此外,Sp、Sn和AUC都有不同程度的提升。所以该预测器可以有效提高识别原核启动子及其强弱类型的性能。
3 结 论
本文引入了一種高效、智能的计算模型iPSI(2L)-XGBoost,通过使用深度学习方法识别原核启动子及其类型。第一层用于识别原核启动子,第二层用于识别启动子是强启动子还是弱启动子。iPSI(2L)-XGBoost融合了One-hot、PCA-PseKNC和PseKNC三种编码方法,并利用卷积神经网络从融合后的One-hot和PseKNC特征编码中提取重要特征,XGBoost用作构建预测器的分类算法。结果表明,与现有方法相比,所提出的智能计算模型的性能显著。所提出的智能计算模型可以有效识别原核启动子和强、弱启动子类型,可能在药物相关应用和学术界具有一定使用价值。
参考文献:
[1] SHAHMURADOV I A,RAZALI R M,BOUGOUFFA S,et al. bTSSfinder: a novel tool for the prediction of promoters in cyanobacteria and Escherichia coli [J].Bioinformatics,2017,33:334-340.
[2] LIU B,YANG F,HUANG D S,et al. iPromoter-2L: a two-layer predictor for identifying promoters and their types by multi-window-based PseKNC [J].Bioinformatics,2017,34(1):33-40.
[3] ABEEL T,SAEYS Y,ROUZÉ P,et al. ProSOM:core promoter prediction based on unsupervised clustering of DNA physical profiles [J].Bioinformatics,2008,24(13):24-31.
[4] MEYSMAN P,COLLADO-VIDES J,MORETT E,et al. Structural Properties of Prokaryotic Promoter Regions Correlate with Functional Features [J/OL].Plos One,2014[2022-10-03].https://doi.org/10.1371/journal.pone.0088717.
[5] WOSTEN M M S M. Eubacterial sigma-factors [J].FEMS Microbiology Reviews,1998,22(3):127-150.
[6] XIAO X,XU Z C,QIU W R,et al. iPSW(2L)-PseKNC:A two-layer predictor for identifying promoters and their strength by hybrid features via pseudo K-tuple nucleotide composition [J].Genomics,2019,111(6):1785-1793.
[7] LE N Q K,YAPP E K Y,NAGASUNDARAM N,et al. Classifying Promoters by Interpreting the Hidden Information of DNA Sequences via Deep Learning and Combination of Continuous FastText N-Grams [J/OL].Frontiers in Bioengineering and Biotechnology,2019,7:[2022-10-03].https://doi.org/10.3389/fbioe.2019.00305.
[8] TAYARA H,TAHIR M,CHONG K T,et al. Identification of prokaryotic promoters and their strength by integrating heterogeneous features [J].Genomics,2020,112(2):1396-1403.
[9] SANTOS-ZAVALETA A,SALGADO H,GAMA-CASTRO S,et al. RegulonDB v 10.5: tackling challenges to unify classic and high throughput knowledge of gene regulation in E. coli K-12 [J].Nucleic Acids Research,2019,47(D1):D212-D220.
[10] FU L M,NIU B F,ZHU Z W,et al. CD-HIT: accelerated for clustering the next-generation sequencing data [J].Bioinformatics,2012,28(23):3150-3152.
[11] CHOU K C. Prediction of signal peptides using scaled window [J].Peptides,2001,22(12):1973-1979.
[12] LE N Q K,YAPP E K Y,HO Q T,et al. iEnhancer-5Step: identifying enhancers using hidden informationof DNA sequences via Chou's 5-step rule and word embedding [J].Anal Biochem,2019,571:53-61.
[13] CHEN W,FENG P M,LIN H,et al. Irspot-psednc: identify recombinationspots with pseudo dinucleotide composition [J/OL].Nucleic Acids Research,2013,41(6):[2022-09-26].https://doi.org/10.1093/nar/gks1450.
[14] XU Y,SHAO X J,WU L Y,et al. iSNO-AAPair:incorporating amino acid pairwise coupling into PseAAC for predicting cysteine S-nitrosylation sites in proteins [J/OL].Peer J,2013[2022-09-26].https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3792191/pdf/peerj-01-171.
[15] BRADLEY A P. The use of the area under the ROC curve in the evaluation of machine learning algorithms [J].Pattern Recognit,1997,30(7):1145-1159.
[16] LIN H,DENG E Z,DING H,et al. iPro54-PseKNC:a sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition [J].Nucleic Acids Research,2014,42(21):12961-12972.
[17] SILVA S D A E,FORTE F,SARTOR I T S,et al. DNA duplex stability as discriminative characteristic for Escherichia coli σ54- and σ28- dependent promoter sequences [J].Biologicals,2014,42(1):22-28.
[18] SONG K. Recognition of prokaryotic promoters based on a novel variable-window Z-curve method [J].Nucleic Acids Research,2012,40(3):963-971.
[19] LI Q Z,LIN H. The recognition and prediction of σ70 promoters in escherichia colik-12 [J].Theoretical Biology,2006,242(1):135-141.
作者簡介:胡仔豪(1999—),男,汉族,江西南昌人,硕士研究生在读,研究方向:生物信息学、智能控制等。
