基于16S rDNA测序的巢湖流域水体粪便污染溯源
2023-06-21祁钊赵相龙桑金慧何振杰傅丹丹岳振宇宋祥军
祁钊,赵相龙,桑金慧,何振杰,傅丹丹,岳振宇,宋祥军*
(1.安徽农业大学信息与计算机学院,合肥 230036;2.安徽省动物性食品质量与生物安全工程实验室,合肥 230036)
环境中的粪便污染是全球范围内日益严重的问题,尤其是在河流和溪流中[1]。流域的粪便污染可能存在多种来源,包括污水基础设施等点源排污,受上游牲畜、宠物和野生动物排泄物污染的径流[2]等。粪便污染带来的高营养和微生物负荷不仅会影响生态健康,而且粪便病原体通过水媒的传播还会进一步影响人类健康。对主要水媒传播病原体的检测是表征人类健康风险的最直接方法,但由于环境中病原种类繁多(包括病毒、细菌和原生动物),对其进行检测不仅会浪费大量时间,还会消耗大量经济成本。为解决这一难题,监管机构使用粪便指示菌(Fecal indicator bacteria,FIB),如大肠杆菌(Escherichia coli)和粪大肠菌群(Fecal coliforms)等来评估粪便污染水平[3],并以此作为水体微生物风险的替代指标[4]。然而,传统粪便指示菌的水质指标检测虽节约了时间和成本,却也存在诸多不足,如无法提供污染源信息、且无法反映水体近期的污染情况[5-6]等,近年来许多污染溯源研究表明,FIB 与环境病原相关性不佳[7-8],故而人们开始探索更加快速、经济的微生物溯源技术。
微生物来源追踪(Microbial source tracking,MST)技术利用人和动物胃肠道中的微生物设计出具有特异性的标记物,并通过定量聚合酶链反应(qPCR)等分子技术检测与宿主相关的微生物标记基因,从而识别潜在的粪便污染源[9-10],但地理差异会显著影响基于宿主特异性分子标记检测方法的灵敏度和特异性,这给MST 技术的实际应用带来了困难。在过去的20 a 中,伴随着下一代测序(Next generation sequencing,NGS)技术的快速发展,研究者对人类、家畜和野生动物肠道微生物组的理解不断加深[11]。使用NGS 数据获取环境及粪便来源的独特的微生物群落图谱的方法被称为基于细菌群落的MST(Community-based MST)[12],主要方法包括基于贝叶斯分类器的Source⁃Tracker[13]与基于快速期望最大化算法的微生物源追踪(FEAST)[14]。两个MST 软件的运行均需要使用者提供粪便来源文库(Fecal taxon library,FTL),并基于该文库中不同的宿主菌群组成对待测样本中微生物的来源进行预测[15],进而评估不同污染源对样品微生物群落的贡献度[16]。SourceTracker 源解析程序通过对不同宿主粪便以及环境水样16S rRNA 基因将不同粪便来源的微生物群落和待分析的水体微生物群落当作一个整体,基于贝叶斯算法,识别水样中不同宿主来源微生物所占比例,解析水样中粪便污染来源[13]。目前,研究人员已将此方法应用在不同区域以识别水环境中不同粪便污染来源及其贡献率,例如,Brown 等[15,17]使用SourceTracker 成功识别出苏必利尔湖河口粪便污染主要来自污水处理厂排放的废水(70%)和海鸥粪便(30%),与qPCR 方法所得结果一致。FEAST是一种新兴的计算工具,可用于同时估计多个潜在来源和各种粪便输入的相对贡献。相同条件下,FEAST比SourceTracker表现出更强的稳定性和更高的运行速度[14]。但目前应用FEAST 软件进行河流水域粪便污染追溯的研究较少,因此其在粪便污染追溯方面的准确性尚需进一步验证。
在本研究中,研究者提取了所采集样本的DNA用于16S rDNA 测序,旨在通过对比测序结果,探索不同采样地点[家禽与废水处理厂(WWTP)、河流水体]及不同样本中(皖江流域地区野生候鸟、人工养殖的家猪)细菌群落结构的多样性,以及不同物种粪便微生物组/环境微生物组的差异。此外,将测得的NGS数据比对到本地潜在病原数据库,用以表征不同生境样本中的潜在病原组成,为后续污染溯源工作奠定基础。而后,通过利用不同来源潜在污染物的NGS 数据集构建FTL 文库,应用基于SourceTracker 与FEAST两种程序的MST 方法对自然水体样本进行污染溯源研究,进一步表征水体公共安全风险,为巢湖流域水环境治理提供理论及数据支持。
1 材料与方法
1.1 研究区域概况
巢湖位于安徽省中部,是中国五大淡水湖之一,同时也是中国富营养化水平最高的湖泊之一[18]。巢湖流域水路网络密集,总流入量的80%以上来自双桥河、派河和南淝河等12 条河流。近年来,伴随巢湖流域人类活动的增加,巢湖受到了严重污染(包括工业、农业和居民生活污水来源),同时逐渐发展出富营养化现象[19]。
1.2 样品采集及处理
1.2.1 水体样本采集
本研究于2021 年10 月采集水体、沉积物样本共63 个,包括从巢湖流域的杭埠河(HR)、丰乐河(FR)、派河(XR)各采集的水样(S)3 份,沉积物3 份,在杭埠河采样点上游有水禽养殖地点(HRD)额外采集的水样3 份。在巢湖北侧农业面源污染监测站(巢湖烔炀镇西宋村污水处理站)采集的处理站入水口(D)及出水口(E)水样各6 份,另外还在监测站周边村庄的排污口(C)采集了6 份水样。另在采集粪便样本时,在淮北昌农收集猪粪便样本(HB)的采样点采集了该养殖厂的废水处理系统进水(ND)以及出水(NC)样本各3份。水样用500 mL无菌塑料瓶收集,沉积物样品使用抓斗取样器在距水面约100 cm 深度处采集。样本于冰上储存,并在6 h 内运送至实验室进行处理。水体样本使用0.22 μm 聚碳酸酯膜进行过滤,所得滤膜储存于-80 ℃以供后续分析。
1.2.2 粪便样本采集
本研究共收集粪便样品229 份,畜禽粪便(猪和鸡)样品取自皖江流域内规模化养殖场。本研究一共采集了6 个不同地点的猪粪便样本,其中包括从太湖县收集的猪粪便样本(PA)、金寨县收集的猪粪便样本(PI)和岳西县收集的猪粪便样本(PL)各5 份,从蚌埠固镇收集的猪粪便样本(BG)、淮北昌农收集的猪粪便样本(HB)、宿州褚兰收集的猪粪便样本(SC)、徐州昌农收集的猪粪便样本(XZ)各4份,在养殖场采集的猪粪便样本共31 份。鸡粪便样本采集自皖江流域内规模化养殖场,共分为5 组,编号为C、mC、L、LS 和S,样本数量分别为9、6、15、15 份及15 份,共60 份。另有鹅粪(F)5 份。所有粪便采集均使用消毒的50 mL收集管,并储存于冰盒,8 h内送至实验室处理。
1.2.3 DNA提取及宏基因组测序
水体、粪便和土壤样本用DNeasy PowerSoil®Pro Kit试剂盒提取DNA,使用通用引物341F(5'-CCTAC⁃GGGNGGCWGCAG-3' ) 和 805R (5'-GACTACH⁃VGGGTATCTAATCC-3')PCR 扩增水中细菌的16S rDNA 基因高变区(V3~V4),PCR 产物经2%琼脂糖凝胶电泳验证。在整个DNA 提取过程中,使用超纯水,以排除假阳性PCR 结果作为阴性对照的可能性。得到的扩增子(PCR产物)经纯化后进行浓度检测,合格后用于测序,扩增子文库的大小和数量分别在Agi⁃lent2100 生物分析仪和Illumina 的文库定量试剂盒上进行评估。
1.2.4 样本构成
为增加数据丰富度及可靠性,除了本研究所采样本外,还使用其他已公布的巢湖水体数据,如Zhang等[20]采集的18份巢湖流域水体样本:具有农业和生活污染背景(Agricultural and domestic pollution,ADPR)的河流包括双桥河(CR1)、拓皋河(CR2、CR12)、鸡裕河(CR3)、兆河(CR10)和玉溪河(CR11);工业和生活污染背景(Industrial and domestic pollution,IDPR)的河流包括南淝河(CR5)、塘西河(CR7)和杭埠河(CR9);农业污染背景(Agricultural pollution,APR)的河流包括烔炀河(CR4)、十五里河(CR6)和派河(CR8);还包括湖中的6个采样点,南淝河口(CL1)、裕溪河(CL6)、兆河(CL3)以及东湖(CL4、CL5)和西湖(CL2)中心的样本。该系列样本采用有机玻璃仪器于水下50 cm深度人工采集,每个地点采集3个平行水样,混合成1个水样,下游处理与本研究相同。此外,本研究还选取了来自菜子湖与升金湖的白额雁(Anser albifrons)粪便样本30 份[21]、升金湖的白头鹤(Grus monacha)粪便样本16份[22]。以及Pan等[23]采集的来自合肥市的87份人类粪便样品。这些样本的下游处理与本研究相同,且测序均为靶向细菌16S rDNA基因V3~V4区。本研究采集水体、粪便样本的测序原始数据已上传至NCBI,并可通过PRJNA783993进行获取。
1.3 数据分析
1.3.1 FTL文库构建
已有研究表明,FTL 文库中是否包含具有明确本地来源的样本对溯源准确度有较大影响,因此本研究选取巢湖流域北侧农业面源污染监测站(巢湖烔阳镇西宋村污水处理站)及周边村庄的排污口样本作为FTL 文库中本地污染来源的代表样品。此外,有研究报道,粪便文库的高组内变异度会对SourceTracker的溯源结果产生显著影响[24]。基于这项原则,本研究将野生水鸟粪便与污水样本按原始样本类型进行了拆分,野生水鸟粪便拆分为白头鹤(Grus monacha)与小白额雁(Anser albifrons)粪便,而所有污水样本按处理前后及污水来源进行了区分,以期最大程度消除粪便文库的组内变异对预测结果产生的影响。
1.3.2 基于16S rDNA测序的潜在病原菌丰度评估
通过VFDB 网站(http://www.mgc.ac.cn/VFs/)以及病理系统资源整合中心(PATRIC)的病原生物数据,以及其他研究中的潜在病原数据[25-29],收集了包括159 个属的潜在病原清单,之后根据属名在美国生物安全协会网站(https://my.absa.org/tiki-index.php?page=Riskgroups)进行检索,记录该属内生物安全等级为二级或三级的物种,于LPSN(The List of Prokary⁃otic names with Standing in Nomenclature)(https://www.bacterio.net/)网站下载该物种的参考16Sr RNA序列,如在LPSN 网站无法检索到该物种,则从NCBI(http://www.ncbi.nlm.nih.gov/)的GenBank中检索并下载对应的16S rDNA 序列。最终构建了包括51 个属,444 个物种16S rDNA 序列的潜在病原数据库。将聚类所得ASV序列通过BLASTN比对到建立的潜在病原数据库,阈值设置为相似度≥98%、覆盖度>99%,潜在病原的相对丰度是通过将鉴定为潜在病原的序列对应的ASV丰度值与样本总ASV丰度的比值来确定。
1.3.3 基于机器学习的微生物污染溯源解析
将不同类型水体或沉积物(即目标样本)设为Sink,微生物污染源或来源的样品(即构建的FTL 文库样本)为Source。分别使用SourceTracker2 以及FEAST 计算来自不同(Source)来源(人类及动物粪便、猪场废水处理系统进水与出水、污水处理厂的进水和出水、村庄污水口)的微生物群落对汇(Sink)环境(即巢湖流域的水体、河流沉积物)的潜在贡献。SourceTracker2与FEAST两种方法的区别在于他们是基于不同的算法来探究目标样本(Sink)中微生物污染源或进行污染来源(Source)的分析。根据Source样本和Sink 样本的群落结构分布,预测Sink 样本中来源于各Source 样本的组成比例。SourceTracker2 与FEAST 的分析使用默认参数进行。每个溯源软件进行5 次独立运行以计算每个潜在源的平均贡献度及其标准偏差。然后通过相对标准偏差(Relative stan⁃dard deviation,RSD,D)计算平均贡献度与标准偏差之间的比率,见公式(1)。
RSD 可以评估多个模型运行的精密程度[30]。式中:S为标准偏差(也可以表示为SD);n为重复次数,xˉ表示平均值。RSD 较高(≥100%)表明预测结果的可靠性低。
Wilcoxon 秩和检验(Wilcoxon rank sum test)用于推断两个独立样本所来自的两个总体分布位置是否有差别,通常在数据不是正态分布时使用。通过Wil⁃coxon 秩和检验可以得到一个正态随机变量Z,再用软件或查正态分布表得到对应的P值。如果P值较小(比如小于或等于给定的显著性水平)则可以拒绝零假设。如果P值较大则没有充分的证据来拒绝零假设,但不意味着接受零假设。即在数据不是正态分布时,可以使用Wilcoxon秩和检验验证样本之间的差异是否显著。
1.3.4 测序数据处理
数据分析参考扩增子分析流程EasyAmplicon(https://github. com/YongxinLiu/Easy Amplicon) 完成[21]。使用USEARCH[22]合并双端序列,去除barcode和引物序列。通过unoise3 去噪获得单碱基精度ASV,基于SILVA 数据库去除嵌合体序列。根据SIL⁃VA 分类器(version2.2,http://sourceforge.net/projects/rdp-classifier/)(置信阈值0.7)获取每个ASV 对应的物种分类信息以及代表序列。设置测序深度阈值为30 000,使用vegan 包进行等量重抽样,通过R(4.0.3)包amplicon(1.11.1)计算Shannon、Simpson 及Chao1指数。通过R 包vegan(2.5.4)计算样本Bray-Curtis 距离以进行PCoA 分析。其余图例均通过OmicStudio tools(https://www.omicstudio.cn/tool)以及R语言进行绘制。
2 结果与分析
2.1 测序数据初步分析
291 个样本(包括228 个粪便样本,63 个水体、沉积物样本)中,共发现10 247 548 条原始序列,经过序列去冗余,获得了73 844 条独特序列,经过unoise3 去噪及基于silva 的去嵌合,聚类生成了包括21 062 条序列的ASV 集合,平均每个样本为412 个ASVs,最低244个ASVs,最高444个ASVs。
2.2 Alpha多样性分析
为了研究数据集样本的微生物多样性,计算了Shannon、Simpson 以及Chao1 指数,总体而言,粪便样本多样性指数水平均低于水体样本。由图1 可见,所有样本中水体沉积物样本具有最高的ASV 数量与物种多样性水平,其次是巢湖水体样本(Wilcoxon 秩和检验,P<0.001),而禽类粪便(包括野生水鸟、鹅与鸡)具有最低的ASV数量与物种多样性水平(Wilcoxon 秩和检验,P<0.01)。

图1 样本alpha多样性指数(log10转换)Figure 1 Differences in alpha diversity indices in all samples(log10 transformed)
2.3 细菌群落结构
经过物种注释,所有样本共注释到31 个门(图2),分布最广泛的门包括变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)、疣微菌门(Verrucomicrobia),其中粪便样本以厚壁菌门为主导(平均相对丰度为55.89%),水体样本则以变形菌门为主导(平均相对丰度为44.13%)。除野生水鸟外,粪便样本门水平细菌集中分布于厚壁菌门、变形菌门与拟杆菌门中,这3 个门平均相对丰度之和占总数的97%以上,而野生水鸟中除这3 种门外,还含有较高相对丰度的放线菌门(平均13.83%)。水体样本门水平细菌更为多样,平均相对丰度在1%以上的有13个门。
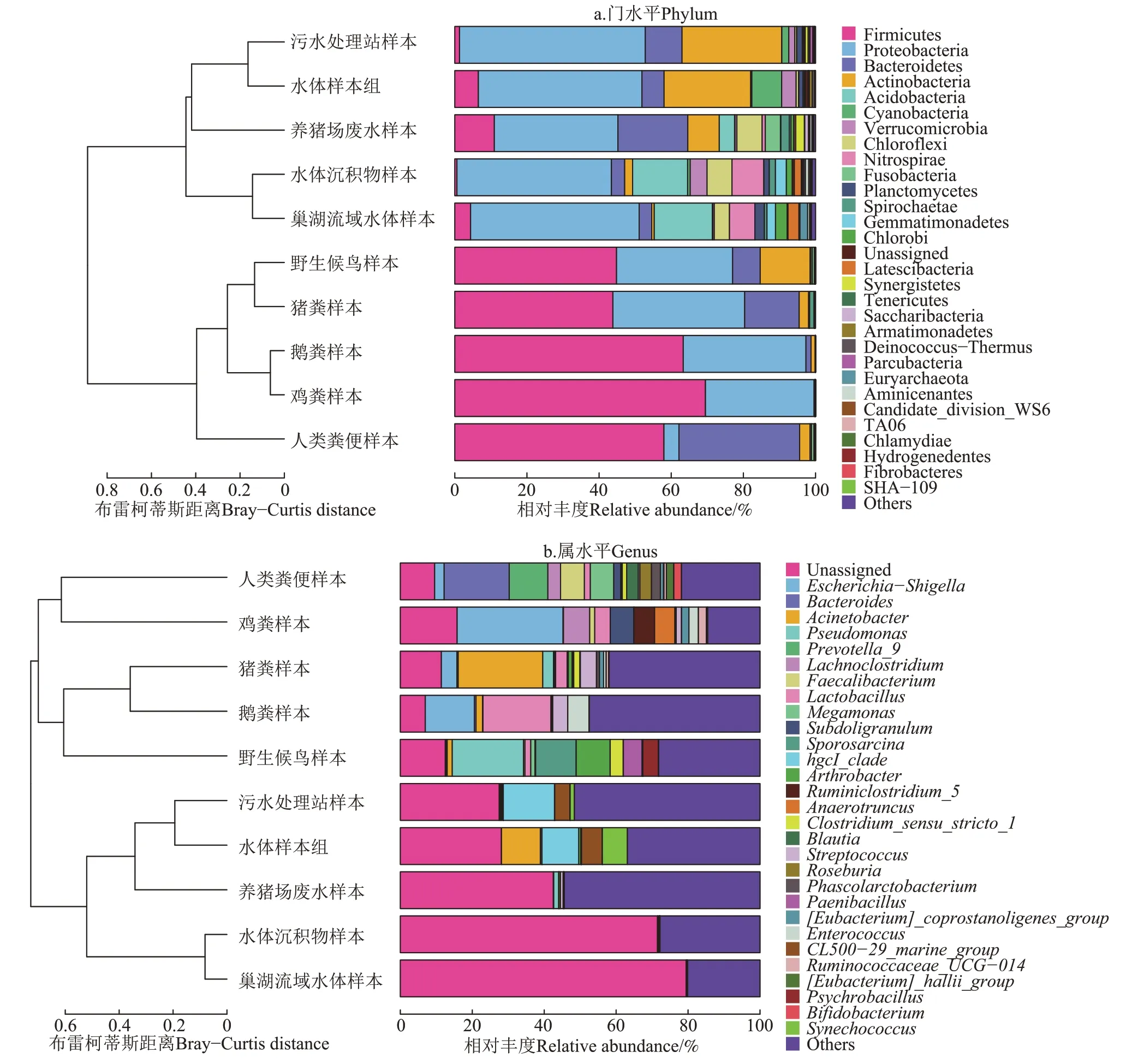
图2 细菌群落(组平均)组成Figure 2 Composition of microbial community
在属水平上,假单胞菌属(Pseudomonas,19.81%)、八叠球菌属(Sporosarcina,11.27%)与节杆菌属(Arthrobacter,9.45%)是野生水鸟中最主要的属,大肠埃氏菌-志贺氏菌属(Escherichia-Shigella,29.46%)是鸡粪中最主要的属,乳杆菌属(Lactobacil⁃lus,18.9%)与大肠埃氏菌-志贺氏菌属(13.64%)在鹅粪中广泛分布,而人粪中表现出拟杆菌属(Bacteroi⁃des,18.09%)与 普 雷 沃 氏 菌 属_9(Prevotella_9,10.74%)的显著富集,猪粪中则以不动杆菌属(Aci⁃netobacter,23.51%)为 主。hgcI_clade在 河 流 水 样(10.23%)与废水样本(14.24%)中表现出明显富集,不动杆菌属表现出在河水中的显著富集(10.78%),此外,河流水样中还发现了较高丰度的CL500-29_marine_group(5.8%)与 蓝 细 菌聚 球藻 属(Syn⁃echococcus,6.96%)。遗憾的是,基于silva 数据库(v123 版本)的注释结果,本研究的河流沉积物与巢湖水样两组样本中分别有71.51%与79.47%的拼接后的测试样本未分类到属。
2.4 Beta多样性分析
基于Bray-Curtis 距离对整体数据进行了主坐标分析(图3),将所有数据分为粪便与水体两个组进行聚类分析。水体样本的beta多样性分析(图3a)表明,猪场废水与污水处理厂废水样本表现出较大的组内变异,相比之下巢湖水体样本和沉积物样本则呈现明显的聚集趋势。粪便样本中,除去各组中少量离群样本,被分类为同一组的样本(野生水鸟、鸡粪、鹅粪、猪粪与人粪)各自均表现出显著聚集(图3b),此外,野生水鸟与猪粪、鹅粪样本表现出明显的聚集,说明其群落组成存在一定相似性。

图3 样本beta多样性差异——主坐标分析Figure 3 Principal coordinates analysis(PCoA)of the microbial community in different groups
2.5 样本中潜在病原的分布
通过将聚类得到的ASV 序列比对到自建病原数据库,对所有样本中潜在病原进行评估,将与数据库的BLAST(相似度≥97%,覆盖度>99%)比对匹配上的病原菌按属水平聚类(图4)。结果显示,所有样本中共注释到57 个潜在病原属,包括145 个潜在病原物种,其中13 个潜在病原属广泛分布于所有样本中,以Pseudescherichia、肠球菌属(Enterococcus)、链球菌属(Streptococcus)与肠杆菌属(Enterobacter)为代表(表1)。在所有样本中,鹅粪、猪场废水与巢湖水样本潜在病原总平均相对丰度最高,分别为0.051%、0.016%与0.011%。从潜在病原数量上来看,含潜在病原属数量最多的为野生水鸟、废水与人粪(分别为48、47、46个属),值得注意的是,猪场废水独有3种潜在病原属,分别为Alloprevotella、福赛坦氏菌(Tannerella)与密螺旋体属(Treponema),而人粪独有一个潜在病原属,即萨特菌属(Sutterella)。
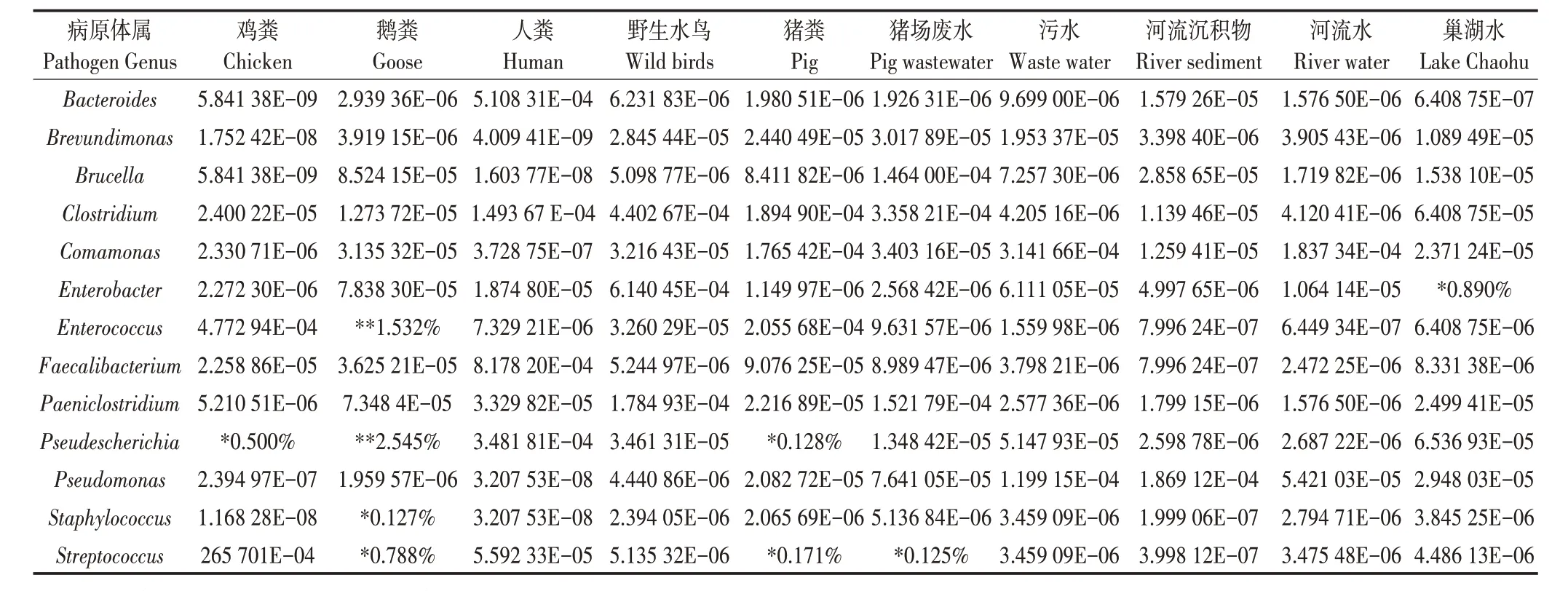
表1 所有样本共有的潜在病原的平均相对丰度(按属聚类)Table 1 Average relative abundance of potential pathogens common to all samples(clustered at the genus level)

图4 潜在病原在样本间的分布(平均相对丰度)Figure 4 Distribution of potential pathogens among all samples(mean relative abundance)
2.6 基于细菌群落的微生物溯源
将不同类型水体或沉积物设为Sink,构建的FTL文库样本作为Source,来判别水体/沉积物样本潜在的污染来源。对于FEAST 以及SourceTracker 两个溯源软件,分别进行5 次独立运行以计算溯源结果的RSD。两个溯源软件对于河流水体污染来源的判定结果较为一致(图5)。

图5 基于NGS数据集的微生物溯源分析Figure 5 Microbial source tracking analysis based on NGS dataset
2.7 FEAST与SourceTracker对比分析
就两个软件预测结果的稳定性(RSD)来看(图6),FEAST的运算结果表现出比SourceTracker更低的RSD 值,其样本平均RDS 值多分布于0.25 及0.50 左右,而SourceTracker 预测值的RSD 计算结果分布在1.0及以上的偏多。

图6 溯源软件5个独立运行结果的相对标准偏差Figure 6 Relative standard deviation generated in 5 independent runs of the traceability software
2.8 微生物溯源结果
FEAST的预测结果表明,沉积物样本的污染来源主要可被划分为猪场废水出水、猪场原始废水以及白头鹤粪便,而相较FEAST 而言,SourceTracker 对于样本污染贡献程度的判定更趋于保守。但综合二者的结果来看,FEAST与SourceTracker溯源软件对于河流水体污染来源的判定结果一致性较高,均将主要的潜在污染来源归类为村庄排污口以及污水处理厂排污口样本,FTL 文库中小白额雁、人类以及鸡粪几乎在所有样本中都不能预测出。对巢湖水污染的预测结果中,两个溯源软件都判定猪场废水出水是主要的污染来源。
3 讨论
研究表明,畜禽肠道定殖的微生物主要有厚壁菌
门(Firmicutes)、拟杆菌门(Bacteroidetes)、变形菌门(Proteobacteria)和放线菌门(Actinobacteria)等,厚壁菌及其家族成员在畜禽粪便样品中具有较高的相对丰度。在所有样本中,水体沉积物样本具有最高的ASV数量与物种多样性水平,而禽类粪便样本具有最低的ASV 数量与物种多样性水平。沉积物作为一个营养丰富的栖息地,为微生物的生长提供了良好条件[31],例如微藻、浮游植物、硅藻、细菌、浮游动物以促进、竞争或共生的方式生活在沉积物上,使水体沉积物发展出丰富的生物多样性[32]。而粪便微生物凭借共生关系存活于宿主肠道,其群落组成受到微生物间互作及宿主的免疫反应等因素影响,并且动物肠道是一个高度厌氧的环境,对粪便微生物群落施加了特定的选择压力,导致粪便微生物群落多样性低于自然水生环境。
在不同类型的水样本中,细菌群落的多样性存在显著差异,巢湖水样的菌群丰度明显高于其他水样,这表明与人类活动相关的较高浓度的有机和无机物可能会降低河流水生细菌群落的物种丰富度,这一结果与之前的研究结果一致[33]。对于研究采集的具有农业和生活污染背景的河流水样而言,物种丰富度和均匀度的降低可能是由于某些物种的高度富集,这些物种很好地适应了有机或无机废水中的特定条件,并且能够通过使用各种营养物质来抵抗或承受环境波动[34]。值得注意的是,有相关研究表明,水体中的粪便污染情况可能会受到季节的影响,不同季节水体中主要的污染源可能会有所差异[35-36]。此外,在对水体样本进行beta多样性差异(主坐标分析)分析时发现,猪场废水与污水处理厂废水样本表现出较大的组内变异,这可能是由于“猪场废水”与“污水”组同时包含了原始废水与处理后废水,而处理过程对细菌群落产生了较大影响,因此导致上述差异。对巢湖水的污染预测结果中,两个溯源软件都判定猪场废水出水是主要的污染来源,但FEAST 预测猪场原始废水贡献了平均2.19%的污染,而在SourceTracker 中仅为0.4%。值得注意的是,CL3样本呈现出与其他巢湖水样不一致的污染预测结果,两个溯源软件均预测其有(23.0±0.5)%的污染来自白头鹤,Zhang等[19]对该采样点的分析指出,该点与其他湖水样本存在明显差异,其含有较高丰度的厚壁菌门(24.13%),而该菌门在其他湖水样本中几乎没有检出。两个溯源软件一致预测存在高水平白头鹤粪便污染的样本还有CR4(SourceTrack⁃er-5.61%与FEAST-9.59%),该样本同样含有较高的厚壁菌门细菌(47.77%),高丰度厚壁菌门细菌的存在或许是该预测结果的主要驱动因素。另外,FEAST的预测结果表明,APR与ADPR背景的河流普遍存在潜在的猪粪污染,污染水平从0.205%(CR3)到4.220%(CR8)。值得注意的是,由于本研究所选污水样本的群落组成在一定程度上可能与粪便样本产生交叉,因此溯源软件对村庄污水处理站以及猪场污水样本较高污染贡献度的预测可能存在一定的误差,存在一定的假阳性率。
对于FEAST 以及SourceTracker 两个溯源软件来说,总体上看,两个基于细菌群落MST 的溯源软件对于巢湖水体以及河流沉积物的预测结果存在较大差异,相较FEAST 而言,SourceTracker 对于FTL 样本污染贡献程度的判定更趋于保守。在FEAST 的预测结果中,沉积物样本的污染来源主要被划分为猪场废水出水、白头鹤粪便以及猪场原始废水,以FR组样本为例,FEAST 预测其污染源平均有18.50%来自猪场废水、8.98%来自白头鹤、4.65%来自猪场原始废水,而SourceTracker 对于FR 组的预测则仅有5.27%来自猪场废水出水、0.49%来自白头鹤、0.81%来自猪场原始废水。在HR 以及XR 组中,FEAST 预测存在3.98%~10.30%的猪场废水出水、3.67%~5.39%的白头鹤以及1.69%~5.40%的猪场原始废水污染,而在Source⁃Tracker 的预测结果中,该比例分别降至0.04%~1.54%、0.08%~0.24%与0.18%~0.70%。
综上所述,尽管基于NGS 的MST 具有众多优势,但其在研究设计方面仍然存在较高要求,后续研究中,存在更少ASV 交叉、地理联系更加紧密的FTL 文库或许能使溯源预测结果更加精准。
4 结论
(1)不同生境样本中,微生物多样性存在明显差异。水样本的微生物群落多样性普遍高于人类及动物粪便样本,其中水体沉积物与湖泊水体样本微生物多样性最丰富。样本/宿主类型是主导细菌群落差异的主要因素,水生环境样本中河流沉积物与污水处理厂细菌群落存在一定相似性,粪便样本中猪粪、野生水鸟与鹅粪存在一定相似性。
(2)变形菌门、放线菌门、拟杆菌门与厚壁菌门在粪便与水体样本中广泛存在,但其相对丰度存在差异。粪便样本以厚壁菌门(平均相对丰度为55.89%)为主,水体样本则以变形菌门(平均相对丰度为44.13%)为主。与本地病原数据库的比对结果显示,以Pseudescherichia、肠球菌属、链球菌属与肠杆菌属为代表的13 种潜在病原在水生环境与粪便样本中广泛分布,这些跨生境样本出现的潜在病原建议在后续研究与监测中给予重点关注。
(3)溯源分析结果表明,河水样本最主要的污染来源是村庄排污口与污水处理厂排污口样本;沉积物与湖水样本则预测出存在猪场排污水与野生水鸟粪便的污染;所有样本未检测到来自人粪与鸡粪的污染。
(4)以自然水体及河流沉积物作为目标样本,溯源软件SourceTracker与FEAST对广泛污染源(包括野生水鸟、家畜、家禽粪便以及生活与养殖业污水)的潜在污染贡献度的预测结果总体相似,但相对同一污染源而言,SourceTracker 对污染贡献比例的判定偏低。总体来看,FEAST的预测结果比SourceTracker拥有更低的RSD值,因此,FEAST模型对潜在污染源的预测结果更具可信度。在实际调研中,推荐结合NGS与传统污染评估方法,以准确探明污染来源,指导水质管理。
