Chromium isotope fractionation during adsorption of chromium(III) by soils and river sediments
2023-06-16ZiyaoFangXiaoqingHeXiYuandLipingQin
Ziyao Fang, Xiaoqing He , Xi Yu, and Liping Qin
1CAS Key Laboratory of Crust-Mantle Materials and Environments, University of Science and Technology of China, Hefei 230026, China;
2CAS Center for Excellence in Comparative Planetology, Hefei 230026, China
Abstract: Chromium (Cr) isotope compositions of sedimentary rocks have been widely used to unravel fluctuations in atmospheric oxygen levels during geologic history.A fundamental framework of this application is that any Cr isotope fractionation in natural environments should be related to the redox transformation of Cr species [Cr(VI) and Cr(III)].However, the behavior of Cr isotopes during non-redox Cr cycling is not yet well understood.Here, we present laboratory experimental results which show that redox-independent adsorption of Cr(III) by natural river sediments and soils can be accompanied by obvious Cr isotope fractionation.The observed Cr isotope fractionation factors (-0.06‰ - -0.95‰, expressed as 103lnα) are much smaller than those caused by redox processes.Combined with previous studies on redoxindependent Cr isotope fractionation induced by ligand-promoted dissolution, we suggest that the systematic shift to highly fractionated Cr isotope compositions of sedimentary rocks is likely to represent atmospheric oxygenation, but muted signals observed in some geologic periods may be attributed to non-redox Cr cycling and should be interpreted with caution.
Keywords: chromium isotopes; adsorption; non-redox Cr cycling; atmospheric oxygen level
1 Introduction
The atmospheric oxygen level fluctuated significantly during geologic history.There was almost no free oxygen in the atmosphere during the first billion years[1].The change in oxygen levels in the atmosphere and oceans is widely thought to have affected the evolution of the biosphere[2,3].
Chromium (Cr) is a redox-sensitive element, and the Cr isotope system has been used to reconstruct the fluctuation of oxygen in geologic history[4-6].In modern surface environments, Cr commonly exists as trivalent Cr [Cr(III)] or hexavalent Cr [Cr(VI)].Chromium in minerals and rocks is mainly composed of highly insoluble Cr(III), while Cr in aquatic environments is dominated by soluble Cr(VI)[7].In natural environments, Cr(III) can be oxidized to Cr(VI)mainly via manganese oxides[8], the presence of which requires free oxygen (see Ref.[9] for an alternative interpretation).Cr(VI) can be reduced by various reductants, such as ferrous iron, sulfides, and organic matters[10-12].Many experimental studies have shown that redox transformation between Cr(VI) and Cr(III) can lead to significant Cr isotope fractionation[5,6].The Cr isotope proxy for atmospheric oxygenation hinges on this redox-induced Cr isotope fractionation.Because igneous rocks have a narrow range of Cr isotope compositions (expressed as δ53Cr; -0.124‰ ± 0.101‰)[13], any divergence of δ53Cr values of sedimentary rocks from this range was proposed to represent the presence of Cr(VI) in the surface environment, further indicating oxidative Cr weathering and then free oxygen in the atmosphere[4].
An assumption in this model is that Cr isotope fractionation should be exclusively linked to redox transformations of Cr.However, Cr can also mobilize in natural environments as Cr(III)[14-16], and studies on possible Cr isotope fractionation during redox-independent processes are still scarce.The nonredox Cr cycling in natural environments consists of three stages (Fig.1): (i) Cr liberation from terrestrial silicate reservoirs through non-redox weathering, (ii) migration of liberated Cr(III) to the ocean, and (iii) incorporation of Cr(III) into marine sediments.Potential Cr isotope fractionation during these stages deserves detailed study, which can help to better understand Cr cycling and the application of the Cr isotope system to the reconstruction of paleoenvironments.For Cr liberation processes, acidic conditions can lead to the dissolution of Cr(III); Cr(III) can also bind to organic ligands andto form complexes and become soluble under natural conditions[15-17].An early experimental study suggested that inorganic acid dissolution of Cr oxides induced no Cr isotope fractionation[18], while subsequent studies illustrated that organic ligand-promoted dissolution of Cr(III) can cause obvious Cr isotope fractionation, with both enriched or depleted53Cr in the solutions[19,20].These results suggest that Cr isotope fractionation in natural environments may not exclusively be linked to redox reactions.Archaea and bacteria,which are thought to have colonized the land since 2.8-2.6 Ga, can secrete organic acids[21]; thus, biogenic organic ligands could be a persistent source of Cr dissolution in geologic history.However, the scale of the biosphere during earlyEarth’s history should be much smaller, so whether organic ligands were abundant enough and played an important role in non-redox Cr cycling is unclear[22].The dissolution of Cr(III) by carbonate ions or inorganic acids might be the dominant process of non-redox Cr weathering in the early geologic period.
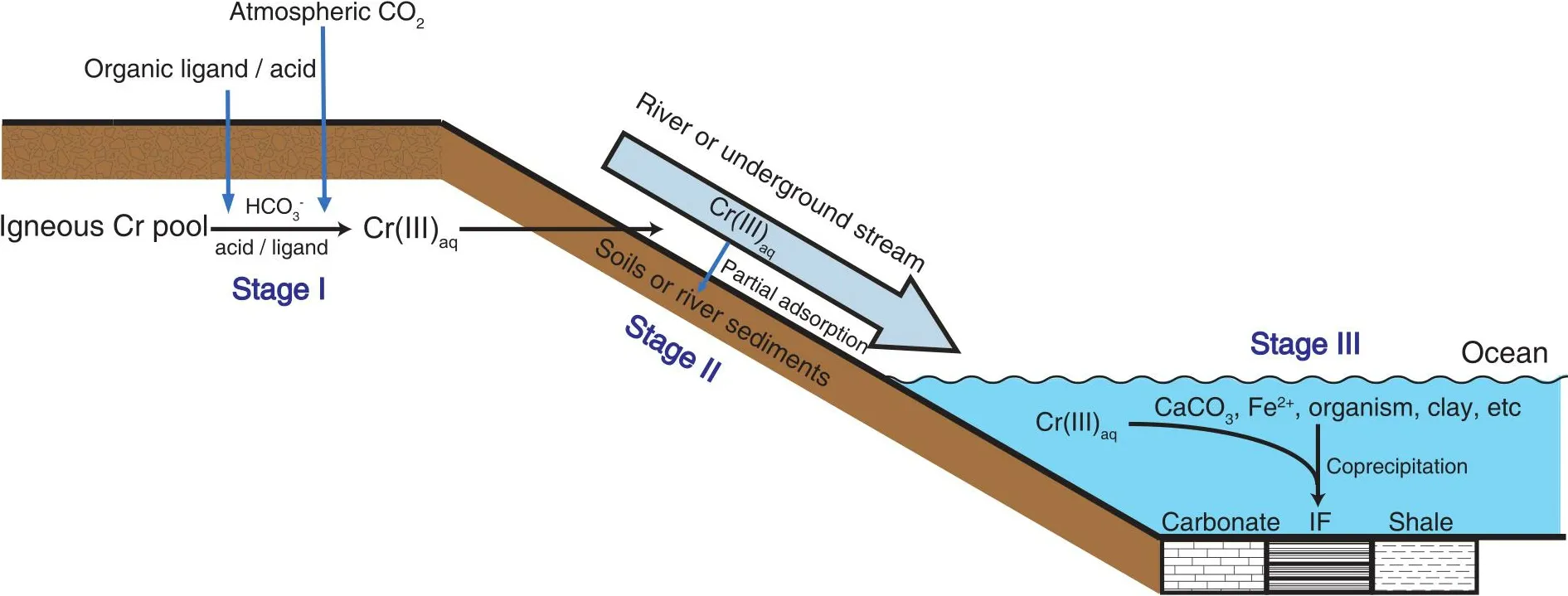
Fig.1.Schematic of non-redox Cr cycling.Stage I: Liberation of Cr(III) in the terrestrial silicate reservoir induced by biogenic organic ligands, acid or.Stage II: The soluble Cr(III) will be partly adsorbed during its transportation in rivers or underground streams.Stage III: Cr(III) in the ocean can be scavenged by multiple processes and preserved in sedimentary rocks such as IFs, carbonates, and shales.
After the liberation of Cr(III) by non-redox weathering, the migration of the dissolved Cr(III) from weathering profiles to the ocean is another process that may induce Cr isotope fractionation.During the migration of Cr(III) to the oceans through underground water, streams, and rivers, the dissolved Cr(III) could be partially adsorbed by soils or river particles.Whether these adsorption processes can cause Cr isotope fractionation is still unclear.Unraveling possible Cr isotope fractionation during adsorption will help to more comprehensively understand the non-redox Cr cycling during the early Earth’s history.Therefore, in this study, a series of experiments were designed to simulate Cr(III) adsorption by soils and river sediments, which are the most common adsorbents in natural environments, and to further investigate Cr isotope fractionation during these processes.
2 Materials and methods
2.1 Materials
Two natural materials were chosen as the sorbents.One was a river sediment sample from the downstream of the Xiaoqing River (37°14.983'N, 118°43.147'E), located in Shandong Province, North China[23].The sample was collected with a stainless steel grab bucket from the surface of the riverbed(0-5 cm).It is rich in organic matters and is dark gray in color.The other material was a red soil sample collected from a paddy field profile in the Yingtan area (28°14'N, 116°53'E),Jiangxi Province, South China.The sample is 60 cm beneath the surface.It is highly weathered and is red in color.The samples were dried at room temperature.The ground sample powders were measured by X-ray diffraction (XRD) to analyse the mineral compositions.Unground samples were used in the adsorption experiments to eliminate the artificial effects on the experiments as much as possible.
Chromium(III) nitrate nonahydrate [Cr(NO3)3·9H2O]powder (99.0%, Sinopharm Chemical Reagent Co.Ltd.) was dissolved in Milli-Q (MQ) water to form a solution with Cr concentrations of 10 ppm and 50 ppm for the adsorption experiments.These solutions were prepared immediately before the experiments.
2.2 Adsorption experiments
Two series of experiments, bottle-incubation and flowthrough, were designed to simulate equilibrium systems and continuous flow systems, respectively (Fig.2).In bottle-incubation experiments, ~5-200 mg of the soil and river sediment samples were first washed with 20 mL MQ water 3 times to remove the possible soluble Cr in the samples.The solids and supernatants were separated by centrifugation, and the supernatants of the last time were collected for Cr concentration analysis to ensure that no Cr in the original samples would be released during the experiments.Then, the adsorbents were mixed with 10 mL of 10 ppm Cr solution and shaken for 24 h in an oscillator.Next, the mixtures were centrifuged, and the supernatants were pipetted out into clean PFA beakers for Cr concentration and isotope composition measurements.In flow-through experiments, ~50-800 mg of the soil and river sediment samples were loaded onto the polypropylene columns, and the columns were then washed with 20 mL MQ water to remove the possible soluble Cr inthe samples.The last 2 mL of eluents was collected for Cr concentration analysis.Next, 2 mL of 50 ppm Cr solution was loaded onto the columns and then rinsed with 2 mL of MQ water for twice.The outlet solutions were collected in clean PFA beakers for Cr concentration and isotope composition measurements.
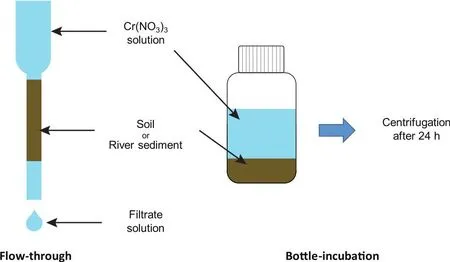
Fig.2.Schematic for flow-through adsorption experiments (left) and bottle-incubation adsorption experiments (right).
2.3 Analytical methods
To characterize the mineral compositions of the adsorbents,the sample powders were analysed by XRD on a Japan Rigaku TTR-III X-ray diffractometer using Cu Kα irradiation(λ= 0.154056 nm).During the measurements, the 2θrange was set to 5°-65° with a step size of 0.02°.
To estimate the possible oxidation of Cr(III) during the experiments, Cr(VI) concentrations in all solutions from both series of experiments were measured by the diphenylcarbazide method with an ultraviolet-visible spectrophotometer(SP-756P, Shanghai Spectrum Instruments Co.Ltd.) at 540 nm, with a detection limit of ~10 ppb.The total Cr concentrations of the solution samples were measured by a Perkin Elmer Elan 6000 inductively coupled plasma mass spectrometer (ICP-MS), with a precision better than 10%.
For Cr isotope analysis, sample solutions containing ~1 μg Cr were mixed with an appropriate amount of50Cr-54Cr double spike and then heated on a hot plate overnight to mix the sample and spike.Double spike was added to correct any mass-dependent Cr isotope fractionation during the whole analysis.Chromium was purified by a two-step cation exchange chromatography procedure.In the first step, the mixtures of the sample and spike were dried and dissolved in 0.2 mL of 6 mol/L HCl and heated at >130 °C.Then, the solution was diluted to 1 mol/L HCl with MQ water and pipetted into a cation exchange column filled with 1 mL Bio-Rad AG 50W-X8 resin (200-400 mesh).Next, 4 mL of 1 mol/L HCl was added to the column.The Cr was eluted as neutral molecules, while other cations were retained in the resin.In the second step, the samples were further purified by another cation exchange column with 0.33 mL Bio-Rad AG 50W-X8 resin.The samples were loaded onto the column in diluted nitric acid.The remaining impurities were eluted by 2.5 mL of 0.5 mol/L HF and 8 mL of 1 mol/L HCl.Then, the Cr was eluted by 3 mL of 2 mol/L HCl.The blank of the whole procedure was <5 ng.The Cr isotope compositions were measured by a Neptune plus multiple-collector inductively coupled plasma-mass spectrometer (MC-ICP-MS).During the measurement, the intensities of four Cr isotopes (50Cr,52Cr,53Cr,54Cr),49Ti,51V, and56Fe were monitored.The latter three were monitored to correct the interferences on50Cr and54Cr.Medium to high resolution mode was used to separate the polyatomic interferences, such as40Ar12C,40Ar14N and40Ar16O.The typical intensity of the52Cr beam was 4-8 V with a Cr concentration of 200 ppb.The spiked internal standard (SCP) and the spiked National Institute of Standards and Technology(NIST) standard reference material (SRM) 3112a were analysed within each analytical session to ensure instrumental accuracy.Each sample was measured twice.The uncertainties for Cr isotope compositions are the largest values among the 2SD of two sample measurements, 2SD of several standard measurements in the same analytical session, and the longterm reproducibility of the standard (0.05‰).
2.4 Notations
Chromium isotope data are expressed as the relative deviation from NIST SRM 979:
The isotope fractionation factorαis used to express the isotope fractionation degree, which is defined as:
whereRrepresents the isotope ratio (53Cr/52Cr).A parameterεis defined to conveniently quantify isotope fractionation:
For equilibrium isotope fractionation, the difference in δ53Cr values between the reactant and the product can be expressed usingΔ:
For kinetic isotope effects, the Rayleigh distillation equation is often used to quantify Cr isotope fractionation, expressed as:
where δ53Cr0and δ53Crtare the Cr isotope compositions of the reactant at the beginning and at timet, respectively, andfis the fraction of the remaining reactant.In the Rayleigh fractionation model,α- 1 (or 103lnα,ε) can be regarded as the isotope fractionation between the remaining reactant and the instantaneous product.
3 Results
3.1 Mineral compositions
Mineralogy results from XRD analyses show that the river sediment sample and the soil sample are mainly composed of quartz and clay minerals (Fig.3).No other crystallized component was detected in the soil sample, while some other minerals, such as albite, pyroxene, and calcite, were identified in the river sediment sample.It is likely that there are other amorphous or poorly crystallized components in the samples,which cannot be easily identified by XRD.Iron-rich minerals,such as ferrihydrite and goethite, are likely to be important components in the red soil, leading to the red color of the sample.The organic phase should also be an important component in the samples, especially in the river sediment sample[23].Almost all these components have previously been shown to have the capacity to adsorb Cr(III)[24-29].

Fig.3.XRD patterns of the soil and river sediment samples.
3.2 Adsorption experiments and Cr isotope compositions
The Cr concentrations of the final washing solutions before the experiments are near the detection limit of ICP-MS, indicating a negligible contribution of indigenous Cr in the river sediment and soil to the adsorption experiments.The fractions of adsorbed Cr(III) by different amounts of solid adsorbents are shown in Table 1 and Fig.4.Both the river sediment and the soil show a large capacity for Cr(III) adsorption.Thefractions of adsorbed Cr(III) by aliquot weight of the adsorbents in the flow-through experiments are less than those in the bottle-incubation experiments (Table 1), which is conceivable, as the adsorption sites of the adsorbents may not be sufficiently occupied during the relatively short time during the flow-through experiments (less than half an hour).No Cr(VI)was detected by the diphenylcarbazide method, suggesting that there was no Cr(III) oxidation or that the Cr(VI) generated by oxidation during the experiments was negligible.
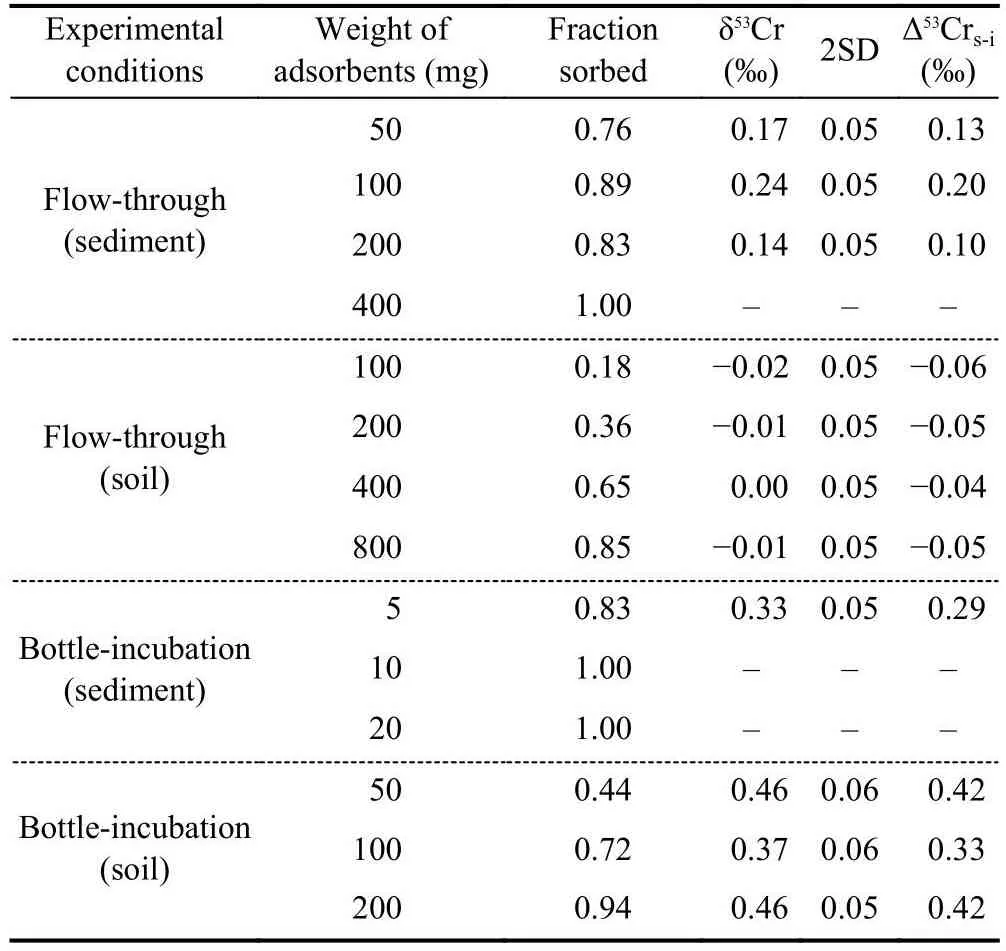
Table 1.The fractions of adsorbed Cr(III) by different amounts of solid adsorbents and the Cr isotope compositions of the remaining Cr after the experiments.Δ53Crs-i means the isotope offset between the remaining Cr and the initial solution (δ53Crsolution - δ53Crinitial).
Because of the large adsorption capacity of the adsorbents,some remaining dissolved Cr(III) in the supernatants are not sufficient for Cr isotope analysis.Four supernatant samples using the river sediment as the adsorbent and seven supernatant samples using the soil as the adsorbent were analysed for their Cr isotope compositions.The results are shown in Table 1 and Fig.4.The remaining dissolved Cr in thesolutions are all characterized by positively fractionated δ53Cr values, except those in the flow-through experiments with soil as the sorbent.The offsets between the δ53Cr values of the remaining dissolved Cr and the initial solution (Δ53Crs-i=δ53Crsolution- δ53Crinitial) in the bottle-incubation experiments are larger than those in the flow-through experiments.In addition, Cr isotope fractionations induced by the soil are larger than those induced by the river sediment in the bottle-incubation experiments.The largest Δ53Crs-ivalue is 0.42‰, observed in the bottle-incubation experiments using the soil as the adsorbent.The Cr isotope data cannot be well fitted by a Rayleigh fractionation model, suggesting that the Cr isotope fractionation during adsorption of Cr(III) by the river sediment and soil samples did not follow a unidirectional Rayleigh distillation process.Moreover, an equilibrium fractionation model also cannot exactly fit the isotope data, indicating that equilibrium status had not been reached during the experiments if the isotope fractionation was caused by isotope exchange between the soluble Cr(III) and the adsorbed Cr(III).Nonetheless, to calculate the range of isotope fractionation factors, we use Rayleigh fractionation and equilibrium fractionation models to fit the individual data points (except the unfractionated data in the bottle-incubation experiments with the soil), and the results show that the isotope fractionation factors (expressed as 103lnα) range from-0.06‰ to -0.95‰ (Fig.4).
4 Discussion
4.1 Cr isotope fractionation during adsorption
Because no Cr(VI) was detected, the positive δ53Cr values of the remaining solution after the experiments should not be caused by redox reactions.Thus, our experimental results suggest that redox-independent adsorption of Cr(III) can be accompanied by Cr isotope fractionation.
The flow-through experiments were designed to simulate the unidirectional adsorption process without much isotope exchange, as the initial solution had flowed through the column in less than half an hour.There are obvious changes in the Cr isotope compositions of the effluents from the flowthrough experiments using the river sediment sample (as least-0.06‰ in 103lnα, Fig.4).A possible mechanism for isotope fractionation is the kinetic effect caused by diffusion.During the flow-through experiments, some Cr(III) in the solution may need to diffuse into the interlamination of the clay minerals before it was adsorbed.Because ions with light isotopes diffuse faster, as a consequence, the solution would be enriched in heavier isotopes.Similar diffusion-induced kinetic Cr isotope fractionation has been previously reported in an experimental study of Cr(VI) adsorption on goethite[30].However, clay minerals are also present in the soil sample(Fig.3), whereas no isotope fractionation was observed in the flow-through experiments using the soil.Therefore, the diffusion process is unlikely to account for the isotope fractionation observed in the experiments, although the different particle sizes of the adsorbents may affect the diffusion process, which needs further specific experiments for evaluation.Instead, the different behavior of Cr isotope fractionation in flow-through experiments using two different adsorbents may be caused by organic matters.The river sediment is enriched in organic matters (TOC value: 0.87%)[23], while the red soil is less enriched (TOC value: ~0.25%)[31].Previous studies suggested that organic ligands can lead to non-redox Cr isotope fractionation; the largest isotope fractionation was observed in small fractions of dissolution of Cr hydroxides by ligands with low metal-ligand stability constants, which may be caused by kinetic effects[20].A similar process may result in Cr isotope fractionation in the flow-through experiments using river sediment in this study, but the intrinsic mechanism is still unclear given that organic ligands can lead to both positive and negative Cr isotope fractionations[20].Alternatively,the observed Cr isotope fractionation in the flow-through experiments using river sediment may be simply attributed to the initial stage of isotope exchange, similar to that in the bottle-incubation experiments, which is discussed as follows.
The observed Cr isotope fractionations in the bottle-incubation experiments are greater than those in the flow-through experiments.The flow-through experiments were all finished within half an hour, while bottle-incubation experiments were conducted for 24 h.We suggest that the adsorption of Cr(III)was initially very quick and quantitative, i.e., nearly all Cr(III) at the initial stage was adsorbed by active adsorption sites; thus, no or very small isotope fractionation was observed at the initial stage of adsorption (such as the flowthrough experiments).Then, isotope exchange between adsorbed Cr(III) and dissolved Cr(III) may have caused Cr isotope fractionation, i.e., equilibrium isotope fractionation.The isotope exchange process may be very slow because isotope equilibrium had not been reached within 24 h in the bottle-incubation experiments with soil; otherwise, the three points in this series should lie on a linear line in Fig.4.
The equilibrium isotope fractionation highly depends on the bonding environments of the element.In our experiments,the soil and river sediment mainly consist of quartz and clay minerals, together with some minor components, such as detrital silicates, carbonate, and probably iron (hydro)oxides and organic matters; all of these components can intensely adsorb Cr(III).Previous studies on the species of Cr(III) adsorption complexes on the surfaces of silica, aluminum (hydro)oxides(γ-Al2O3,γ-AlOOH), clay minerals (kaolinite, illite, and montmorillonite), calcite, and alfalfa biomass all showed that Cr(III)mainly forms a monodentate or bidentate inner-sphere complex with dimers or polymers of Cr—O octahedra[24,25,27,29,32,33].As dissolved Cr(III) in solution is predominantly monomeric Cr—O octahedra[32,34], the polymerization of Cr—O octahedra and chelation with surface functional groups during adsorption may lead to a slight change in the bond energy around Cr, resulting in Cr isotope fractionation during isotope exchange (equilibrium isotope fractionation).Because the specific coordination shells around Cr on the surface of various adsorbents can be slightly different, such as the atom types in the second coordination shell, the equilibrium isotope fractionation factors during adsorption of Cr(III) may not be identical, which may account for the differences we observed in experiments using the soil and the river sediment as the adsorbents.On the other hand, because the first coordination shells around Cr in both the monomer and polymer complexes are composed of 6 oxygen atoms, the differences in bond energy should be relatively small compared to that between Cr(VI) and Cr(III).Therefore, the equilibrium isotope fractionation factor should also be smaller than that between Cr(VI) and Cr(III)[35].Specific studies of Cr(III) adsorption by isolated components in soil and sediment are needed to further understand the mechanism of Cr isotope fractionation.
The experiments in this study are only simply designed, so they cannot represent all circumstances in natural environments.In natural non-redox Cr cycling, Cr(OH)3(aq), Cr(III)-carbonate complexes, and Cr(III)-organic ligand complexes may be more common dissolved species.Further adsorption experiments using these species as adsorbates are needed to comprehensively understand Cr isotope fractionation during the transportation of Cr(III) in non-redox Cr cycling.
4.2 Implications for Cr isotopes as a proxy for atmospheric oxygenation
Previous studies have shown that organic ligand-induced nonredox dissolution of Cr(III) can cause Cr isotope fractionation[19,20].Whether this process played an important role during the early Earth’s history with only a small scale of the biosphere is unclear.The experiments in this study show that adsorption of Cr(III) by natural river sediments and soils can be accompanied by obvious Cr isotope fractionation, further suggesting that the migration process of dissolved Cr(III) during non-redox Cr(III) cycling in natural environments can also lead to positively fractionated δ53Cr values.Thus, deviation of δ53Cr values observed in sedimentary rocks away from the igneous range should not necessarily correspond to the presence of Cr(VI) and then a high atmospheric oxygen level.
Nonetheless, it is noteworthy that the isotope effects of redox and non-redox Cr cycling are distinctive.The experimentally determined equilibrium isotope fractionation factor between Cr(VI) and Cr(III) is ~ -5.8‰ (103lnα)[35], and the values for kinetic isotope fractionation range from -2‰ to-4‰ (103lnα)[6], much greater than the values caused by adsorption (103lnα= -0.06‰ - -0.95‰) in this study.Although the calculated isotope fractionation factors may not be accurate enough because of the non-equilibrium conditions in this study, the intrinsic equilibrium isotope fractionation factors are likely to be small due to the little change in bonding environments around Cr during adsorption (see discussion in Section 4.1).Moreover, the large isotope fractionations induced by ligand-promoted dissolution were mainly observed in small fractions of dissolution by low stability constant ligands, which may indicate that the overall ligandpromoted isotope fractionation in natural environments should be much smaller (< 1.3‰, Δ53Crsolution-solid)[20].Hence,we infer that the significantly positively fractionated Cr isotope composition observed in some sediments can be readily explained by oxidative weathering.In contrast, some muted positively fractionated δ53Cr values in sedimentary rocks may not be related to an increase in atmospheric oxygen level(“whiff of oxygen”), as previously suggested[4,18,22], because the suboxic surface environment at the onset of atmospheric oxygenation would result in significant partial reduction of Cr(VI) during transportation and the remaining Cr(VI) in the oceans should be characterized by extremely positive δ53Cr values instead of by muted signals.Although we are not able to set a threshold of δ53Cr values to represent the onset of oxidative Cr weathering, it may be helpful to use the boxplot of δ53Cr values in different periods to recognize the signals from oxidative weathering (Fig.5).The occasional slightly fractionated δ53Cr values caused by non-redox Cr cycling under a reducing atmosphere would be plotted as outliers in the boxplot if there are sufficient data points.In this light, the slightly positively fractionated δ53Cr values observed in sedimentary rocks from the Archean and possibly some periods in the Proterozoic were likely caused by non-redox Cr cycling and should not reflect transient atmospheric oxygenation events.

Fig.5.(a) Compilation of Cr isotope compositions of sedimentary rocks throughout Earth history from the literature[4, 18, 22, 36-80].The gray band indicates the range for igneous reservoir[13].(b) Boxplot of the Cr isotope data at 100 million-year intervals.The box comprises the 25th and 75th percentiles, the black square in the box denotes the mean value, the line in the box represents the median value, the whiskers are the 2.5th and 97.5th percentiles, and the hollow dots are outliers.
5 Conclusions
This study reports Cr isotope fractionation during the adsorption of Cr(III) by natural soils and river sediments.The mainfindings are as follows:
(Ⅰ) The adsorption of Cr(III) by soils and river sediments,with no redox transformation of Cr, can be accompanied by obvious Cr isotope fractionation (103lnα= -0.06‰ - -0.95‰).Thus, fractionated δ53Cr values out of the igneous range should not necessarily be interpreted by redox cycling of Cr.
(Ⅱ) As the magnitudes of Cr isotope fractionation caused by non-redox Cr cycling are much smaller than those in redox reactions, the significantly positively fractionated Cr isotope composition of some sedimentary rocks can be explained by atmospheric oxygenation, but some muted signals may be caused by non-redox reactions.
Acknowledgements
The authors thank the anonymous reviewers for their constructive comments.This work was supported by the National Key R&D Program of China (2021YFA0718200), the National Natural Science Foundation of China (42103006,42103007), the Pre-research Project on Civil Aerospace Technologies (D020202) of the Chinese National Space Administration, the Natural Science Foundation of Anhui Province (2108085QD163), and the Fundamental Research Funds for the Central Universities of China (WK3410000019,WK2080000152, WK2080000154).
Conflict of interest
The authors declare that they have no conflicts of interest.
Biographies
Ziyao Fangis an Associate Research Fellow at the University of Science and Technology of China (USTC).He received his Ph.D.degree from USTC in 2020.His research interests include metal stable isotope geochemistry, paleoenvironment reconstruction, and astrobiology.
Xiaoqing Heis a Postdoctoral Researcher at the University of Science and Technology of China (USTC).She received her Ph.D.degree in Geology from USTC in 2020.Her research interests include metal stable isotope geochemistry, Earth’s surface processes, and paleoenvironment reconstruction.
杂志排行

中国科学技术大学学报的其它文章
- 中文概要
- Harnack inequality for polyharmonic equations
- Research on the influence of social relations on social enterprise performance: The mediating role of business model innovation
- E-commerce cart recommendation effects: A field experiment on entertainment products
- New criteria of supersolubility of finite groups
- Diagenetic effects on strontium isotope (87Sr/86Sr) and elemental(Sr, Mn, and Fe) signatures of Late Ordovician carbonates