小鼠miR-155基因启动子分析及转录活性鉴定
2023-06-15罗朦莎李雪琴张莺莺
罗朦莎,李雪琴,张莺莺,吕 坤
巨噬细胞作为一种极具可塑性和多能性的细胞群体,在不同生理和病理状态下,主要极化为两种类型:M1型即经典活化的巨噬细胞,具有促炎作用;M2型即替代性活化的巨噬细胞,具有抗炎症作用[1-4]。微小RNA(microRNA,miRNA)是一类广泛存在于真核细胞中的长度为19~24个核苷酸的非编码小分子调控RNA,在基因调控方面起着极为重要的作用,控制细胞的生长发育,决定细胞分化的类型,调节细胞增殖与凋亡[5-8]。miR-155是一个已知能影响巨噬细胞M1/M2分化的miRNA分子[4,9-13]。本课题组前期研究发现,在巨噬细胞极化过程中miR-155表达失调,且高表达于M1型巨噬细胞。然而,目前对于巨噬细胞中miR-155表达失调的转录调节机制仍知之甚少。本研究通过构建连续截短的miR-155基因启动子荧光素酶报告基因质粒,初步鉴定miR-155基因启动子转录活性区域和潜在的转录因子,为阐明miR-155基因在巨噬细胞极化中表达失调的转录调控奠定基础。
1 材料与方法
1.1 材料 BALB/c小鼠10只,6~8周龄,体质量22 g左右,由南京青龙山实验动物中心提供。质粒pLMP、pGL3-Basic、pGL3-promoter及pRL-TK(Promega公司);大肠杆菌感受态细胞DH5α(TianGen公司));人胚肾上皮细胞(HEK293T细胞)、小鼠成纤维细胞(L929细胞)由皖南医学院弋矶山医院中心实验室提供。质粒提取试剂盒、基因组提取试剂盒、DNA凝胶回收试剂盒(Axygen公司);Q5 High-Fidelity DNA聚合酶、限制性内切酶XhoⅠ、HindⅢ、BamHⅠ、T4 DNA连接酶(NEB公司);dNTP、转染脂质体Lipofectamine2000(Invitrogen公司);qRT-PCR试剂盒(Qiagen公司);RPMI 1640培养基(HyClone公司)、DMEM培养基(HyClone公司)、胰蛋白酶(碧云天公司)、胎牛血清(FBS,Gibco公司);双荧光报告基因检测试剂盒(Promega公司);所有引物均由深圳市华安平康生物科技有限公司合成;测序由深圳市华安平康生物科技有限公司完成。
1.2 方法
1.2.1 引物的设计与合成 从miRBase(http://www.mirbase.org/)获得小鼠miR-155前体序列。取miR-155上游2 000 nt作为其启动子区,利用引物设计软件Primer Premier 5.0,设计相应的PCR扩增引物。miR-155前体引物命名为miR-155-F、miR-155-R,上、下游引物分别引入内切酶XhoⅠ和BamHⅠ酶切位点;miR-155启动子引物为:miR-155-p-F、miR-155-p-F1、miR-155-p-F2、miR-155-p-R。其中miR-155-p-R为通用下游引物,分别获得miR-155上游2 000 nt、1 000 nt和500 nt的片段。上、下游引物分别引入内切酶XhoⅠ和HindⅢ酶切位点及保护碱基(见表1)。
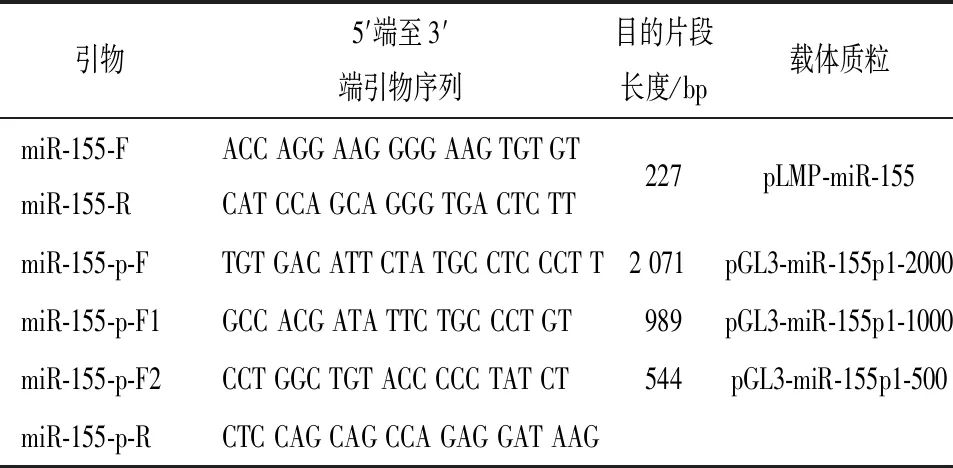
表1 扩增miR-155前体及miR-155启动子基因片段的引物序列
1.2.2 小鼠骨髓来源巨噬细胞(bone marrow-derived macrophages,BMDMs)的分离与培养 小鼠颈椎脱臼处死后,无菌分离股骨和胫骨,用L929细胞条件培养基(含20%FBS和20% L929细胞培养3 d上清的DMEM培养基)冲洗骨髓内容物并制成细胞悬液。将细胞悬液均匀点种于6孔板中,在37 ℃和5% CO2的条件下培养7 d 即为BMDMs。
1.2.3 miR-155前体及miR-155启动子系列片段的PCR扩增 提取BMDMs细胞基因组DNA作为模板,PCR扩增miR-155前体片段和启动子片段。对于miR-155基因启动子系列截短片段的扩增,以构建成功的pGL3-Basic-m-miR-155质粒为模板,分别用相应的上游引物和下游引物配对扩增,合成截短片段。反应结束后,PCR产物通过1%琼脂糖凝胶电泳检测,对获得的单一条带进行凝胶回收。
1.2.4 miR-155前体及启动子系列截短片段与荧光素酶报告基因载体的重组 miR-155前体及miR-155启动子系列截短片段均采用无增强子和启动子,只有一个Luc基因的pLMP和pGL3-Basic作为报告载体。分别用限制性内切酶XhoⅠ、BamHⅠ和XhoⅠ、HindⅢ对回收的PCR产物和pLMP、pGL3-Basic载体质粒进行双酶切,37 ℃酶切温育1 h,直接凝胶回收酶切产物。用T4 DNA连接酶将酶切回收后的两片段进行连接,16 ℃孵育过夜。将连接产物转化至大肠杆菌感受态细胞DH5α、涂板、挑菌,经菌落PCR鉴定DNA大小正确后,对样品提取质粒,再分别用XhoⅠ、BamHⅠ和XhoⅠ、HindⅢ进行双酶切,电泳鉴定重组质粒DNA大小正确后,将正确的菌液送公司测序,测序结果比对,将miR-155前体命名为:pLMP-m-miR-155。miR-155启动子系列缺失片段命名为:pGL3-Basic-m-miR-155、pGL3-Basic-m-miR-155-1、pGL3-Basic-m-miR-155-2。
1.2.5 重组质粒转染及双荧光素酶报告分析 293T细胞用含10% FBS的DMEM培养基培养。在转染前1 d将293T细胞以5×105个/毫升接种于12孔板中,37 ℃、5% CO2培养箱中培养,直至细胞密度约达60%。按照Lipofectamine2000试剂盒操作说明,将miRNA表达载体(无关microRNA Rno-miR-344及不含miRNA的空载体作为负对照)与小鼠miR-155启动子荧光素酶报告载体(以不含启动子序列的空载体pGL3-basic作为负对照)瞬时共转染293T细胞,48 h后裂解细胞,用试剂盒检测双荧光素酶活性,验证mmu-miR-155启动子区域转录活性。
1.2.6 与miR-155启动子结合的转录因子的预测 取mmu-mir-155上游2 000 nt作为其启动子区,将miR-155启动子序列输入TransFac数据库中的MATCH软件,根据网站提示进行操作,输出可以与miR-155 promoter序列结合的转录因子的预测结果。
1.2.7 与miR-155启动子结合的转录因子在M1型和M2型巨噬细胞中的表达水平检测 按1.2.2的方法制备获得小鼠BMDMs细胞,培养7 d后换为含10% 胎牛血清的RPMI-1640培养基继续培养1 d,用终浓度为100 ng/mL 脂多糖及20 ng/mL γ-干扰素刺激8 h极化为M1,用20 ng/mL白细胞介素-4刺激24 h极化为M2。收集细胞,用Trizol提取细胞总RNA并逆转录成cDNA,Q-PCR检测筛选出来的转录因子mRNA的表达。Q-PCR反应步骤如下:95 ℃预变性3 min,95 ℃变性30 s,60 ℃退火30 s,重复40个循环,进行熔融曲线分析。用2-ΔΔCT法测定mRNA或miRNA的表达水平,并分别与GAPDH或U6水平进行归一化,每个样本3个复孔。
1.3 统计学方法 采用t检验、方差分析和q检验。
2 结果
2.1 miR-155前体及miR-155启动子系列片段的PCR扩增和重组荧光素酶报告基因质粒的构建 以小鼠BMDMs细胞基因组为模板,PCR扩增小鼠miR-155基因前体及启动子系列片段(见图1)。将miR-155基因前体PCR产物插入到pLMP载体中,而miR-155基因启动子系列片段PCR产物插入到pGL3-Basic载体中。再以构建好的重组质粒为模板,PCR扩增相应的片段,并对PCR产物进行测序验证。经测序对比,克隆序列正确,符合设计要求。表明已成功构建miR-155前体及miR-155启动子系列截短片段的报告基因质粒。
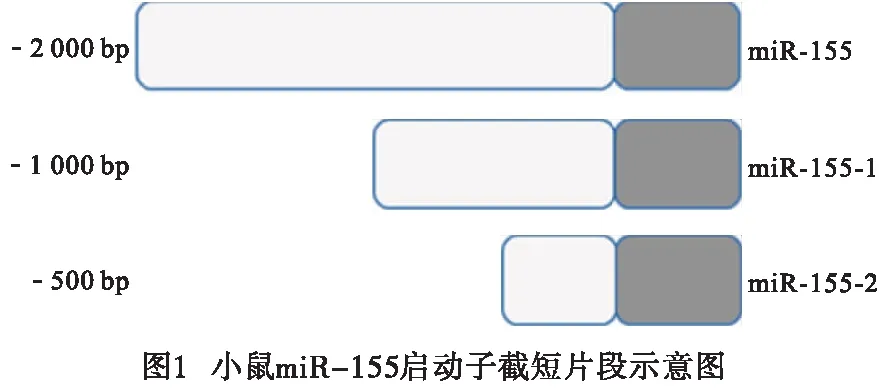
2.2 小鼠miR-155启动子双荧光素酶检测 将小鼠miR-155启动子系列片段报告基因质粒转染293T细胞,48 h后采用双荧光素酶报告检测系统检测转录活性,以无关microRNA Rno-miR-344及不含miRNA的空载体作为负对照,将阴性对照pGL3-Basic的荧光素酶相对活性值设为1。结果显示,miR-155上游1~500 bp、1~1 000 bp及1~2 000 bp片段均存在转录激活作用,且miR-155上游1~500 bp的转录活性最高(P<0.05)(见表2)。

表2 小鼠miR-155启动子双荧光素酶检测结果

表2
2.3 与miR-155启动子结合的转录因子的预测分析 为了探索小鼠miR-155的上下游调控关系元件,我们提取miR-155基因转录起始位点(TSS)上游2 000 nt序列作为其启动子区域,预测结合该区域的转录因子(TF)数据。使用转录因子数据库和转录因子结合位点预测程序完成转录因子预测分析。TF预测方案及结果转录因子数据库使用TRANSFAC 7.0 public。转录因子结合位点预测使用pwmatch程序,设置log-odds的cutoff值。结果显示,在小鼠miR-155基因TSS上游2 000 nt区域存在Crp、Pax-6、Elf-1、Gata-1、Hnf-4、BR-C Z4等转录因子结合位点(见表3)。
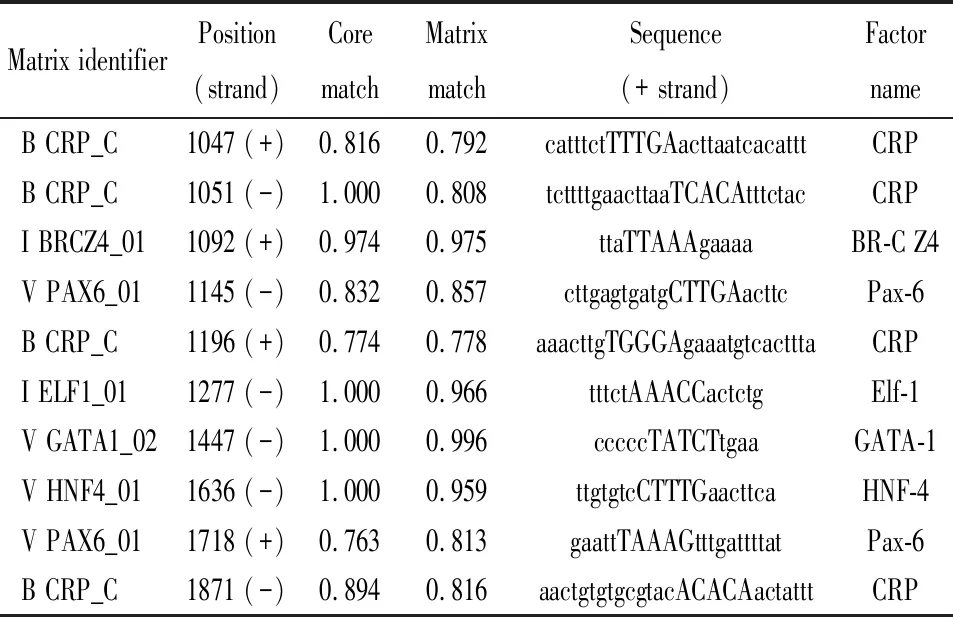
表3 结合小鼠miR-155启动子的转录因子预测结果
2.4 结合miR-155启动子的转录因子在巨噬细胞极化过程中的表达分析 根据上述预测结果,我们选取其中4个match分数最高的转录因子Crp、Pax-6、Elf-1和Gata-1,检测其在体外极化的M1型和M2型巨噬细胞中的表达。RT-qPCR结果显示,体外诱导的M1型和M2型巨噬细胞极化成功,iNOS高表达于M1型巨噬细胞而Arg1高表达于M2型巨噬细胞中(P<0.01);Crp、Pax-6、Elf-1和Gata-1 4个转录因子均在M1型巨噬细胞中低表达,与M1型巨噬细胞中高表达的miR-155的表达方式相反(P<0.01)(见表4)。
3 讨论
miRNA是一类非编码蛋白质的内源性单链小分子RNA,主要在转录后水平上调控基因表达。目前大部分研究主要关注筛选差异表达的miRNA及其调控的靶基因,而对它们自身的转录调控研究并不多。已知大部分miRNA的表达都是通过RNA聚合酶Ⅱ指导进行转录的,而RNA聚合酶Ⅱ指导的转录常常受到转录因子的调控,提示miRNA的转录过程也可能受到转录因子或者信号通路的调节。有研究[5-6]报道miRNA的表达可能受到相关转录因子的调控,如miR-217通过影响JAK 3/STAT 3信号通路来抑制M2型巨噬细胞进而调控卵巢癌的发生与转移[5]。现有的研究[11-14]表明,转录调节在miR-155的表达调控过程中发挥重要作用,如miR-155能有效抑制M1巨噬细胞中iNOS和TNFα基因的表达,同时有助于抑制其有效的mRNA靶点Inpp5d、Tspan14、Ptprj和Mafb等[4]。然而,目前对于巨噬细胞中miR-155表达失调的转录调控机制仍知之甚少。
表4 RT-qPCR检测M1型和M2型巨噬细胞中各因子的mRNA表达水平
本研究通过构建连续截短的miR-155基因启动子荧光素酶报告基因质粒,成功克隆得到3条不同长度的截短片段(即miR-155基因TSS上游1~500 bp、1~1 000 bp及1~2 000 bp)。双荧光素酶活性实验证实,miR-155基因TSS上游1~500 bp,1~1 000 bp及1~2 000 bp区域均存在转录激活作用,提示miR-155基因TSS上游1~500 bp区域很可能为miR-155基因启动子核心区域。接着,利用TransFac数据库中的MATCH软件预测,得到可能结合miR-155基因TSS上游1~2 000 bp这一区域的转录因子,并选取其中结合评分最高的4个转录因子Crp、Pax-6、Elf-1和Gata-1在小鼠M1型和M2型巨噬细胞中进行差异表达分析,结果显示这4种转录因子均在M1型巨噬细胞中低表达,且在M2型巨噬细胞中高表达。同时,本研究又对M1型和M2型巨噬细胞中miR-155的表达水平进行检测,发现miR-155在M1型巨噬细胞中高表达,在M2型巨噬细胞中低表达,这与前期研究[8,15]相符。因此,推测上述4个转录因子可能参与miR-155转录表达的负性调节,本研究为进一步揭示miR-155表达失调的转录调控机制奠定基础。
