Role of p53 suppression in the pathogenesis of hepatocellular carcinoma
2023-06-15HeenaChoudharySatishMandlikDeepaMandlik
Heena B Choudhary,Satish K Mandlik, Deepa S Mandlik
Heena B Choudhary, Deepa S Mandlik, Department of Pharmacology, BVDU, Poona College of Pharmacy, Pune 411038, Maharashtra, India
Satish K Mandlik, Department of Pharmaceutics, BVDU, Poona College of Pharmacy, Pune 411038, Maharashtra, India
Abstract
Key Words: Hepatocellular carcinoma; p53; Tumour suppressor gene; Murine double minute 2; Chemoresistance
INTRODUCTION
Hepatocellular carcinoma (HCC) is the most prevalent form of primary liver cancer and typically brought on by chronic liver diseases. HCC every year has 841000 new cases and 781000 deaths which makes HCC the fourth leading cause of cancer-related death globally[1]. By 2025 the annual diagnosed cases of HCC are expected to outstretch up to 1 million. HCC can appear on its own or more usually results from chronic liver illnesses of various etiological factors, like cirrhosis, alcoholic steatohepatitis(ASH) and non-ASH (NASH), viral infections [hepatitis C virus (HCV) and hepatitis B virus (HBV)] and hepatitis B and C[2] (Figure 1). As transplantation, ablation and resection can only be used for the treatment of early-stage HCC patients and since the majority of patients are diagnosed with severe stages or intrinsic chronic hepatic disease, discovering novel therapies to increase survival time and lower the likelihood of tumour recurrence is an important objective in the domain of HCC. According to the Barcelona clinic liver cancer staging system, Sorafenib has been utilized for several years as a treatment for HCC that has progressed to an advanced stage[2]. Nevertheless, concerns regarding its effectiveness and harmful effects have been raised[3], including the activation of metabolizing enzymes[4]. While the use of immunotherapy has altered the approach to treating advanced HCC, not all patients with HCC respond positively to these recent therapies. Consequently, there are ongoing investigations into various strategies to enhance the effectiveness of HCC medication by incorporating chemosensitizers[5] and/or personalized combination treatments[6]. Additionally, patients who receive immunotherapy may experience an upsurge in immune-related adverse events[7]. In this situation,tailoring HCC therapy to each patient is becoming an essential priority in order to achieve the greatest possible clinical advantage. Along with this, there is a need to find new biomarkers that can identify potential responders to pharmacological treatment by accurately predicting the clinical response[2].
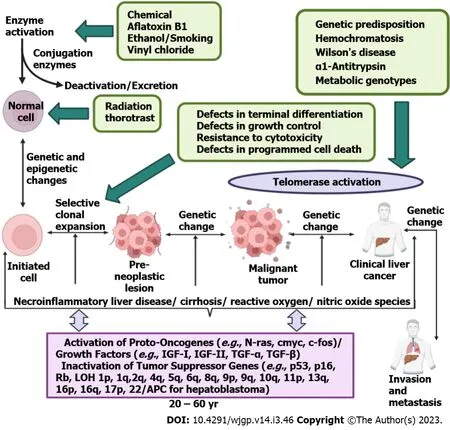
Figure 1 Etiological factors of hepatocellular carcinoma. Aflatoxin B1, vinyl chloride, smoking, ethanol, and many such factors leads to the development of hepatocellular carcinoma (HCC). Genetic predisposition, hemochromatosis and metabolic genotypes could also be the reason for the initiation of the HCC. There are many stages in the development of HCC including formation of pre-neoplastic lesion and malignant tumour (Created with BioRender.com). IGF: Insulin-like growth factor; TGF-α: Tumour growth factor-α; TGF-β: Tumour growth factor-β.
The wild-type protein of the tumour suppressor gene p53 is responsible for regulating the cell cycle and inducing apoptosis after DNA damage[8]. However, if p53 undergoes a mutation, cells with DNA damage can avoid apoptosis and develop into cancerous cells[9]. Additionally, the mutant p53 protein,which loses the function of the wild-type protein, can amass in the nuclei of cells and is considered to be a highly specific marker for malignancy[10]. Studies have indicated that alterations in p53 are linked to tumour differentiation, vascular invasion, tumour stage, Child-Pugh class and serum alpha fetoprotein in HCC[11]. Nevertheless, the prognostic significance of p53 alterations in HCC is not yet fully understood and conflicting results have been reported in various studies[8]. While some studies have suggested that tumour p53 upregulation and serum anti-p53 antibody elevation are linked to recurrence-free survival and overall survival in HCC patients, other studies have not confirmed these findings[12]. Furthermore, most studies have indicated that HCC patients with a mutant p53 phenotype experience poor survival, but the prognostic effect has varied widely with a broad range of hazard ratios due to small and diverse studies[13].
Despite the progress made in treating certain other types of cancer, there isn’t any effective treatment available for HCC and presently, chemotherapy and surgical resection are the two most common treatment options[14]. As a result, learning more about the molecular basis for the etiology of HCC may help researchers to find novel therapeutic approaches[14]. This review seeks to examine the biological mechanisms implicated in development of HCC, with a specific focus upon the murine double minute 2(MDM2)-p53 networks and the hypothesised function of MDM2 in suppressing p53 expression. By knowing how MDM2 contributes in the development of HCC, we may potentially find methods to inhibit HCC as well as discover novel therapeutic approaches[14]. We also explore the function of cellular biology in identifying novel HCC biomarkers, utilizing MDM2-p53 linkages as prognostic indicators and considering future treatment strategies for HCC[14].
TUMOUR SUPPRESSOR P53
Nuclear protein that is produced by the human p53 gene is essential for cell division. It takes up roughly 20 kb of DNA. It was located on the short arm of chromosome 17 (17 p. 13.1)viasouthern filter hybridization of DNA from a hybrid of human-rodent[15]. There are 11 exons in the gene. The human p53 protein has 4 primary functional domains and 393 amino acids, according to earlier studies. There are more discussions of the p53 gene's and protein's molecular structure in the literature[16]. The discovery that Simian virus 40 (SV40)-transformed cells produce novel protein type which might be extracted by serum of anti-SV40 tumour led to the detection of the p53 protein, a protein having a noticeable molecular mass of 53 kDa[17]. Additional research showed that fluorophenylalanine therapy of SV40-infected monkey cells resulted in an increment in cellular thymidine absorption and a progressive suppression of viral and host DNA synthesis[14]. Transformed mouse cells, such as leukemias, murine sarcoma virus and spontaneously transformed fibroblasts, have been shown to express p53 in various cellular studies[18,19].In-vitrovirus sequence-specific DNA linkage is blocked by the wild-type p53 protein's complex formation with the hepatitis B virus X protein (HBx). The broad range of p53 function changes caused by HBx contributes to the emergence of human HCC[20]. The discovery of p53 in human leukaemia cells and transformed cell lines raises the possibility that p53 might influence the gene makeup of some types of leukaemia[21]. The production of p53 protein seems towards being essential for the mechanism of physiological cycling cells, in addition to its control over how dormant normal cells move from the G0-phase to the S-phase of the cell cycle. While p53 in experimentally altered (Meth A) mice cells appeared to have entered the stage of translation of RNA, transcription of p53 in growth inhibited healthy cells is regulated at the transcriptional or post-transcriptional stage[22]. Additionally,p53 takes involvement in the conversion of healthy embryonic cells and can work alongside Ha-ras oncogene to accomplish this[23]. In untransformed cells, it displayed a short half-life of 5 to 10 min.
Previous research has indicated that p53 can alter normal fibroblasts so that they become tumourbearing cells[24]. However, later research revealed that dominant-negative mutants were present in a conserved area of p53 in the initially cloned p53 cDNAs utilised in the earlier experiments. Its biological processes and conformation were improved by these changes. This idea was supported by research done in the late 1980s, which also demonstrated that the wild-type p53 functioned as a tumoursuppressing gene and prevented transformation of oncogene[14]. Also, it was demonstrated that the p53 gene commonly had mutations in human large intestine cancer. These results and subsequent research demonstrated that p53 functioned as a tumour-suppressing gene. In reality, numerous investigations have supported the widely accepted theory that authentic wild-type p53 functions as a tumoursuppressing gene and exhibits certain wild-type p53 features. The key points from p53 study in this field could be described in the following way:
First, a mutant p53 gene combined with RAS activation can prevent transformation. Second, the p53 gene mutation is the most frequent gene change found in human cancers. There are regional differences in the p53 mutational profile[25] and disruption of normal p53 activity frequently promotes cancer inhibition and advancement[26]. Finally, mutant p53 proteins might function as suppressive proteins that decrease the action of wild-type p53 proteins by competing with them[27]. Additionally, MDM2-p53 binding can be blocked by modified p53, which kills tumour cells that overexpress MDM2. The combined approach for the destruction of MDM2-p53 connection with radiation revealed improved anticancer effectsin-vitroandin-vivo, as proven by Koomet al[28]. As a transcription factor, p53 exhibits tumour-suppressing propertiesviacontrolling the transcription of genes[14]. p53 has a shallow level of activation in healthy cells. A variety of stress signals, such as DNA damage, oncogene activation and hypoxia, are detected by p53[14]. As a result, in response to stress signals, p53 is overexpressed and accumulates in high quantities. The expression of the relevant genes is then transcriptionally regulated by p53 after it attaches to a particular DNA sequence known as the p53 responsive element (RE) in the DNA. The cellular roles of p53 gene have been identified through studies, including regulation of cell cycle, apoptosis, senescence, DNA repair, alteration of metabolism and metastasis[29,30]. The interplay of certain p53 functions with particular oncoproteins, like MDM2. The following sections of this review go into further detail about these interactions. Posttranscriptional changes primarily regulate the levels of the p53 protein. Ubiquitination, phosphorylation, acetylation, methylation and SUMOylation are a few of these alterations. Ubiquitination is the primary modification that controls p53 in comparison to other changes[31] and a variety of deubiquitinating enzymes control p53 stability[32].
MDM2, a negative regulator of p53, is the primary controller of p53[14]. MDM2 participates in this mechanism by directly interacting with p53 and mediating its ubiquitination-dependent destruction(Figure 2). Fourth, a molecular similarity of MDM2, that attaches to the p53 transcriptional activation region and blocks it from activating transcription, has been discovered in the MDM4 (MDMX) protein[14]. MDM4 thereby aids in the development and operation of tumours. It's essential to note that MDM4 can prevent the breakdown of the MDM2 protein by interacting with the MDM2 protein through the RING finger domain, controlling the function of MDM2 in suppressing p53 activity. As a result, both MDM4 and MDM2 are regularly upregulated in several types of human malignancies. Schlaegeret al[33] have demonstrated that MDM4 and EFF1A2 could function as etiology-independent oncogenes in a sizable portion of human HCC.
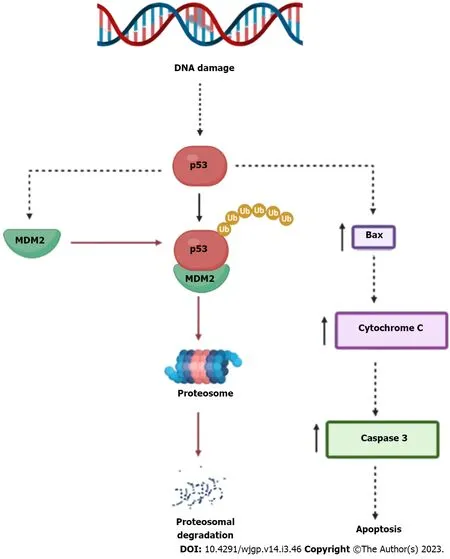
Figure 2 Regulation of p53. p53 is a tumour suppressor protein which leads to increase in the apoptosis markers like Bax, cytochrome C and caspase 3. As murine double minute 2 complexes with p53 it leads to ubiquitination of p53 and proteasomal degradation takes place (Created with BioRender.com). MDM2: Murine double minute 2.
PATHOGENESIS OF P53-ASSOCIATED HCC
The mechanisms described in this section, which may also apply to other cancer types, have been shown to promote the progression of HCC by changing the MDM2-p53 pathway[34]. As numerous ribosomal proteins (RP) that link to MDM2 are secreted from the nucleolus when ribosome biogenesis is hindered,it has already been demonstrated that p53 stabilized by ribosome biogenesis disruption[35]. There is further proof that a decrease in rRNA synthesis can stabilize p53 by blocking MDM2-mediated p53 damage by binding to RPs released from the nucleolus in the regenerating rat liver following partial hepatectomy[34,36]. It has been established that the p53-binding HBx, which localizes p53 to the cytoplasm, is crucial for the growth of HCC[34]. In cells that possess the HBx protein, doxorubicin therapy has been found to raise p53 levels; in addition, doxorubicin therapy recovers p53-mediated transcriptional function by lowering MDM2 amounts and raising p53 nuclear accumulation[34,37].Similar to this, the tumour suppressor KLF6 has been found to be inactivated in response to HCV infection, and KLF6 expression has been linked to a poor prognosis for HCC[38]. The Kruppel-like €C2H2 zinc finger family, which has been linked to cell differentiation, signal transduction, and cell cycle control, includes KLF6[34]. According to Tarocchi and colleagues, reduced KLF6 expression results in increased MDM2 and decreased p53, and these disparities in the MDM2-p53 pathway is additionally linked to reduced survival in patients with surgically removed HCC[34]. In contrast, increased KLF6 expression decreases MDM2 expression and increases p53 expression in HCC cell lines[39]. In order to boost MDM2 ubiquitin ligase activity and p53 degradation, Junget al[40] discovered the Enigma LIM domain protein, which plays a role in signal transductionviaprotein kinases[34,40].
Serum response factor (SRF), which is likewise elevated in HCC and promotes additional MDM2 stabilization and p53 degradation, can also trigger Enigma[40,41]. By causing MDM2 phosphorylationviaAkt signalling, Minand and colleagues demonstrated that one p53 target gene, phosphatase of regenerating liver 1 (PRL-1), which is overexpressed in a number of cancersviaan unknown mechanism, firmly downregulated p53 levels and inhibited p53-mediated apoptosis, forming yet another feedback loop that contributes to HCC development[34]. According to research, Inhibitor of Growth-1 is a type II tumour suppressor that influences cell activity by changing the chromatin's structure and controlling transcription[42]. According to research by Menget al[34], ING1 suppresses the growth of hepatoma cells by causing apoptosis and cell-cycle arrest[34]. Both an increase in p14ARF expression that inhibits MDM2 and an increase in p53 acetylation and activation are likely responsible for these tumour suppressor functions[34]. It has been shown that iron status affects p53 activity by suppressing MDM2 expression and that this reduction in MDM2 expression prevents the development of HCC[43].Because p53 has been demonstrated to be linked to growth arrest by lowering iron absorption and intracellular iron concentration through interaction with iron-RE-binding proteins, there may be a feedback loop between p53 stimulation and iron concentration[34]. Sirtuin-3 (Sirt3) is a member of the mammalian sirtuin family that is found in the mitochondria and helps to regulate metabolic activity. It is also linked to the Warburg effect, which is the dysregulation of cancer cell metabolism[44,45].
In addition, Sirt3 overexpression reduced HCC cell growth and promoted apoptosis in HepG2 and HuH-7 cell lines by upregulating p53 protein activity, which is found to be downregulated in human HCC tissue[46]. The balance between MDM2 and p53 is thrown off in HCC[34]. Each of the elements demonstrated has a distinct part in promoting the growth of cancer, although defective p53 is invariably identified as the primary cause of HCC[34]. To provide a better understanding of these pathways and other as-yet-unidentified mechanisms that contribute to HCC formation through the modification of p53, further systematic and thorough research is required[34].
ROLE OF P53 IN MECHANISMS OF HEPATOCARCINOGENESIS
Hepatocarcinogenesis is a multi-step process that includes stages of initiation, promotion and advancement. It is characterized by the collection of genetic and epigenetic alterations. Proto-oncogene and tumour suppressor gene genetic changes are highly significant in the development of human cancer,according to molecular genetic studies[47]. In fact, the development of HCC is attributed to over 20 genes linked in 4 carcinogenic pathways[48]. These numerous genetic changes appear to be linked to the development of tumours and multistep carcinogenesis. During chemically generated HCC in rodents,stimulation of oncogenes from Ras family and other families was also discovered; however, it is minimal proof of comparable stimulation in solid cancers[49]; changes in oncogenes have indeed been identified in only a tiny portion of human tumours[50]. Contrarily, there’s proof that tumoursuppressing genes, including ones for p53, p16INKa and pRb, are changed at various phases of hepatocarcinogenesis, which may either directly or indirectly lead to chromosomal destabilization and encourage cell growth and neovascularization[51,52]. A variety of human cancer forms, including HCC,have been shown to frequently exhibit mutations in the residual genotype and deletion of one genotype of the p53 tumour-suppressing gene[53,54] (Figure 3).
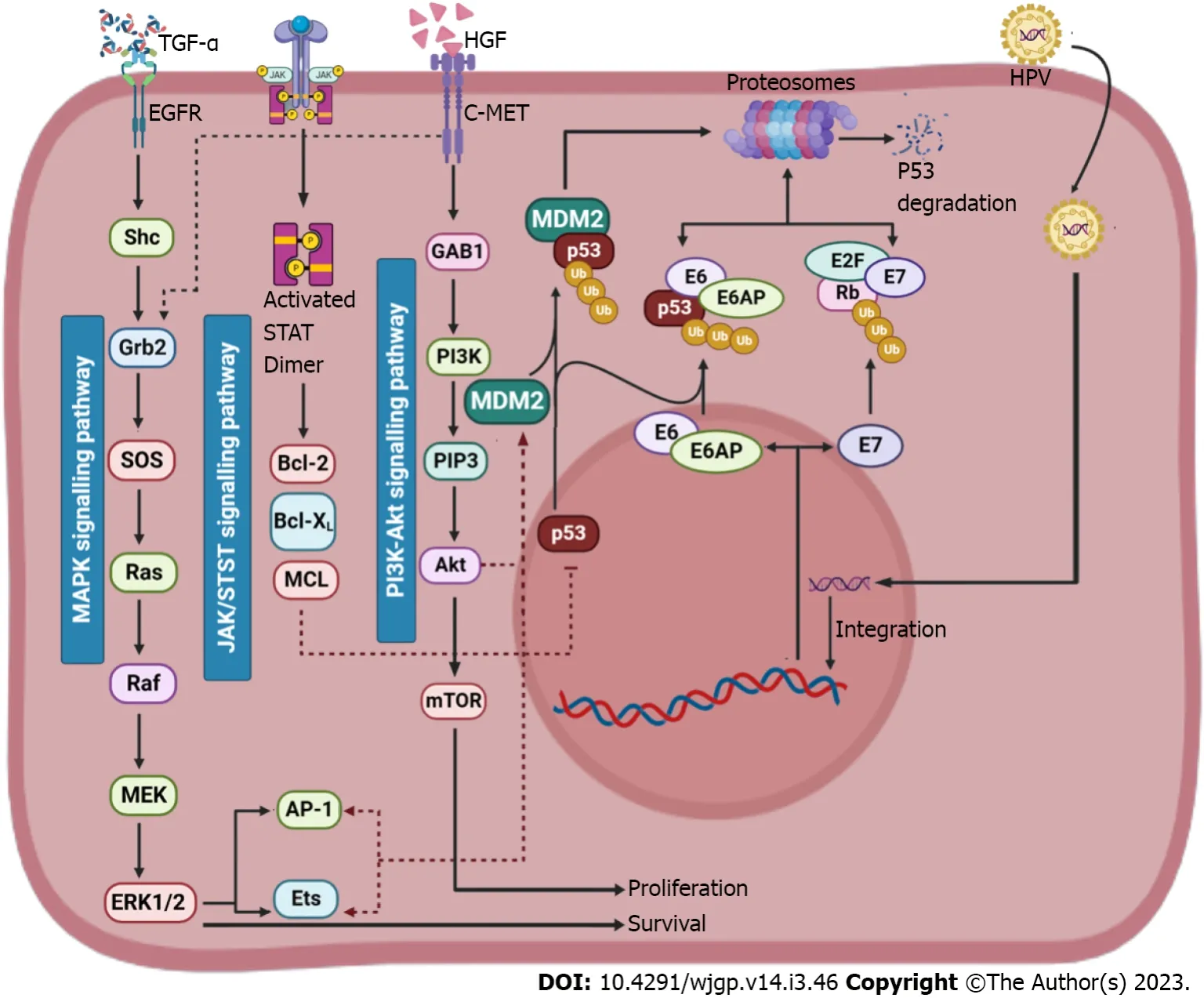
Figure 3 Role of p53 in pathogenesis of hepatocellular carcinoma. Murine double minute 2 (MDM2) suppresses the tumour suppressing activity of p53 by formation of a MDM2-p53 complex. The activation of mitogen-activated protein kinases and PI3K-Akt signalling pathway activates the production of MDM2 and the activation of JAK/STAT signalling pathway suppresses the p53 protein (Created with BioRender.com). TGF: Tumour growth factor; HGF: Hepatocyte growth factor;EGFR: Epidermal growth factor receptor; HPV: Human papillomavirus; MAPK: Mitogen-activated protein kinases; MDM2: Murine double minute 2; MEK: Mitogenactivated extracellular signal-regulated kinas; ERK1/2: Extracellular signal-regulated kinase 1/2; mTOR: Mechanistic target of rapamycin; AP-1: Activating protein-1;MCL: Medial collateral ligament.
p53 function
The p53 protein is known to act as a transcriptional factor, however, it may still be discovered to have additional roles[55-57]. It induces the transcription of several genes implicated in the management of the cell cycle, such as the growth arrest and DNA damage-inducible gene GADD45[58], the gene encoding p21WAF1/CIP1 (also known as p21), the gene encoding MDM2 (a negative regulator of p53) and the gene encoding 14-3-3 (a regulator of G2-M progression)[59]. Additionally, it stimulates a number of genes that are likely involved in apoptosis, such as the gene for Bax, a pro-apoptotic protein linked to Bcl-2 and a number of genes that encode proteins included in the production of reactive oxygen species(ROS)[55,57]. The regulation of the cell cycle, DNA synthesis and repair, cell differentiation, transcriptional repression, genomic plasticity and apoptosis have all been linked to p53[53,60]. This includes stopping the cell cycle at G1 and G2 to evaluate genomic integrity. It is crucial for the detection of DNA damage and the subsequent reaction[61]. Cells express more p53 when their DNA is compromised. The cell can then activate the DNA repair systems because this protein prevents the G1 phase. p53 overexpression triggers apoptosis when the damage is too severe to be repaired, which results in cell death[62,63].
p53 regulation
Numerous levels of strict regulation are in place for the p53 protein. Protein activity, protein stability and subcellular distribution are recognized as the three main levels of control[61]. Recently, thorough reviews on additional p53 regulation topics, like p53 post-translational changes and how they affect p53 activities, have been reported[64]. The cell cycle regulates how the p53 protein moves among the cytoplasm and nuclear compartments[65]. For p53 to function as a tumour suppressor, it must build up in the nucleus. An effective way for tumour cells to remain to multiply in the existence of wild-type p53 is to avoid nuclear aggregation[61]. In fact, cytoplasmic sequestration of p53 has frequently been seen in a variety of tumours, including neuroblastomas, breast cancer and colon cancer[66,67]. MDM2 is the factor that causes p53 to accumulate in the cytoplasm of at least some of these tumours and other proteins have also been linked to cytoplasmic retention[68-70]. Many viral proteins, including the human papillomavirus E6 protein[71], the adenoviral E1B 55 kDa protein and the HBV HBx protein[72,73] affect the location of p53. Additionally, several tumour types have indeed been linked to p53 import/export abnormalities[74]. p53 is a protein that shuttles back and forth through the nuclear pore complex. A bipartite nuclear localization signal (NLS) found at the C-terminal domain of p53 and two nuclear export signals found in the N- and C-terminal areas are responsible for nucleocytoplasmic transport. NLS mutations impede MDM2-mediated cytoplasmic breakdown and inhibit p53 export[75].
p53 alterations and hepatocarcinogenesis
A damaged p53 gene, which is typically rendered inactiveviaa single mutation or a gene deletion, is implicated in about 50% of all malignancies. These modifications are thought to hinder p53 from oligomerizing and forming the tetrameric complexes which link to particular DNA sequences, changing the way the wild-type protein behaves physiologically[53,63,76]. Human malignancies with a p53 mutation are more deadly, aggressive and likely to metastasize. Therefore, p53 abnormality detection may reveal details about the molecular pathogenesis and -etiology of human cancer[77,78].
The two most common genetic changes associated with human HCC are mutations of p53 gene and HBV DNA incorporation into the host gene. An abnormality in the p53 gene has been found in 30%-60%of HCC patients[79] and a hotspot of mutations in the gene has been identified in HCC patients from locations with significant aflatoxin exposure[80]. Aflatoxin B1, which contaminates foods in regions where it is endemic, clearly plays a part in the development of hepatocarcinogenesis because its metabolites encourage the formation of apurinic sites and G to T mutations in chromosomal DNA and the presence of a codon 249 G to T transversion in p53 in 50% of HCC patients from high aflatoxin exposure areas[80].
The transactivation domain of wild-type p53 is polymorphic at residue 72, where a single-base mutation results in the substitution of proline for arginine (CCCCGC)[81]. Numerous research has been conducted on the prevalence of these two alleles and their potential association with the chances of developing cancer, even though the clinical significance of the variants is unknown[61]. According to one study, hepatocarcinogenesis may be influenced by the frequent loss of the proline allele in HCVassociated liver cancer. To clarify this claim, however, further research on this topic has to be done[81].Furthermore, Okadaet al[82] discovered a strong association between HCV type 1b infection and male homozygotes for p53Pro. Therefore, there may be a connection between this p53 polymorphism and HCV infection.
ROLES OF P53 IN EXTRINSIC FACTOR-INDUCED LIVER CARCINOGENESIS
Aflatoxin B1
Humans can consume aflatoxin B1 (AFB1), a very known mutagen of the liver made by theAspergillus fungus, through contaminated food products[83,84]. According to one research, the population inhabitants’ risk of HCC caused by AFB1 is 17% in some regions of the world[85]. According to the mechanism, CYP40s convert AFB1 into AFB1-8,9-epoxide, which then interacts with DNA to generate 8,9-dihydro-8-(N7-guanyl)-9-hydroxyaflatoxin B1 (AFB1-N7-guanine) adducts[86]. If these adducts are not repaired, they cause G>T transversions to occur while DNA replication[87,88]. The DNA binding domain of p53 is known to be altered by an arginine to serine missense mutation at codon 249 (R249S),that is brought on by a G>T transversion at codon 249's third base[89]. About 50% of HCC cases in places where AFB1 Levels are high, like Qidong City in China, contain the p53 R249S mutation[90],suggesting that p53 might have a role in AFB1-induced HCC[86].
In addition to the guanines at codon 248, AFB1-8,9-epoxide combine with the p53 gene, although those guanine adducts need not produce cancer-inducing alterations as often as p53 R249S[88,89,91].While p53 is first activated by AFB1-mediated DNA damage to cause cell cycle arrest during the S to G2/M phases[92], liver cells which acquire p53 R249S will indeed bypass this cellular defense process with a preferential benefit for proliferation, that might further progress towards liver cancer[86]. In fact,p53R249S is demonstrated to boost insulin-like growth factor 2 (IGF-2) transcription in Hep3B (p53 null)cells, indicating a potential gain-of-function activity[86]. IGF-2 has been linked to accelerating the course of human HCC and is overexpressed in 16%-40% of cases[93]. Additionally, there is a strong association between the genotype IGF-2 +3580 AA and the threat of HCC. It's interesting to note that HepG2 cell viability and proliferation are reduced when IGF-2 is silenced[86]. As a result, p53 mutation caused by AFB1 is critical for the development of HCC, presumably through increased IGF-2 signalling[86].
Vinyl chloride
It is debatable whether vinyl chloride (VC), a carcinogenic gas involved in the production of polyvinyl chloride, can cause human HCC, but it primarily causes angiosarcomas of the liver (ASL)[94-96]. The liver's CYP2E1 enzyme converts VC from the lungs into chloroethylene oxide, which causes liver cancer by forming large DNA adducts[97,98]. 7-(2-oxoethyl)-deoxyguanosine, 3,N4-etheno-deoxycytidine,1,N6-ethenodeoxyadenosine and N2,3-etheno-deoxyguanosine are four VC-associated DNA adducts that have been identifiedin-vivo[97].
According to reports, the p53 gene's codons 179, 249 and 255 had more A>T transversions in VCinduced human ASLs[99,100]. According to a study employing Sprague Dawley rats, the most of p53 mutations in HCC and ASL after exposure of VC, are A>T transversions; the A>T transversions in ASLs are found at 253 codons of rat p53, that is equal to 255 codons in humans[86,101]. A conformationspecific p53 antibody, PAb240 and other p53 antibodies have been used to identify an increase in the quantities of mutant p53 protein in serum samples from workers exposed to VC[99,102].
Alcohol
Drinking alcohol regularly is bad for our health. It causes liver cirrhosis, which then develops into HCC.One of the major causes of HCC is alcoholic liver disease[103,104]. Alcohol usage is linked to an up to twofold increased risk of HCC, say case studies from throughout the globe[105,106]. Studies on mice on an alcohol diet have also revealed exacerbation of inflammation, epithelial-mesenchymal transition(EMT), fibrosis, as well as progression to HCC[105]. However, hazardous by-products of alcohol catabolism, such as a buildup of acetaldehyde and free radicals, might affect oxidative stress, apoptotic cell death, necrosis, and necroptosis[106]. Pure ethanol cannot directly induce inflammation and liver damage[106]. Elevated inflammatory cytokine release brought on by ongoing inflammatory pathways leads to the creation of ROS[103,106]. HCC growth is aided by ROS-induced DNA damage, hepatocyte genomic susceptibility, and T-lymphocyte suppression[103].
Additionally, alcohol catabolism affects a number of lipid metabolism processes, which results in liver steatosis and fatty acid oxidation inhibition[105,106].
An essential biological process known as epigenetic reversibility of gene expression frequently contributes to cancer[106]. Alcohol abuse alters epigenetic pathways that result in altered DNA methylation and acetylation. For instance, aberrant acetylation has been linked to alcohol-induced HCC and hepatic steatosis[106]. In a syngeneic BALB/c mouse tumour model, promoter hypomethylation in circulating tumour cells of HCC is strongly correlated with overexpression of c-Met and hepatocyte growth factor[107].
Alcohol consumption also affects metabolism, causing acetaldehyde to accumulate, hypomethylation,an absence of antioxidants and retinoic acid, as well as inflammation, oxidative stress, hypoxia, and genetic instability[106].
Non-alcoholic fatty liver disease
NASH, non-alcoholic fatty liver disease (NAFLD), fibrosis, cirrhosis and HCC are among the illnesses that make up NAFLD. Obesity, type 2 diabetes and metabolic syndrome are all linked to NAFLD[108].NAFLD affects 20%-30% of people in the West, according to estimates[109]. Just 11.5% of individuals with NAFLD-caused cirrhosis go on to grow HCC and roughly 50% of NASH-caused HCCs happen in people who don't have cirrhosis[110,111]. These findings suggest that additional oncogenic events are necessary for NAFLD-caused HCC[86]. The cellular pathways in NAFLD caused HCC, however,remain poorly known. Its development has been linked to a number of mediators, including insulin resistance, deregulation of NF-kB signalling, the Pl3K/ATK-PTEN pathway and the activity of certain miRNAs (e.g., miR-34)[112,113].
Additionally, p53 has been linked to the development of NAFLDviaa variety of pathways. In a mouse model for NAFLD, in which p53 +/+ and p53 -/- mice have nourished a diet deficient in choline and methionine, p53 +/+ mice exhibit enhancement in histologically visible steatohepatitis, fibrosis and ROS formation as well as higher protein levels of p66Shc, a protein linked to oxidative stress, in comparison to p53 Human NASH hepatocytes have increased p53 activity along with elevated p21 and p66Shc mRNA levels, that is highly linked to the degree of fibrosis[86]. Additionally, a rise in miR-34 expression and reduced SIRT1 protein content were found in human NAFLD liver tissues along with a rise in acetylation of p53, both of that were associated with the condition[114]. By promoting hepatocyte death, stimulation of the miR-34a-SIRT1-p53 axis has also been demonstrated to lead to liver fibrosis or NASH[115]. Additionally, p53 has the ability to activate miR-34, which suppresses the expression of SIRT1 mRNA and raises p53's acetylation in a positive feedback loop[116]. These findings suggest that NAFLD and high expression of miR-34 and p53 are related. However, it should be highlighted that p53-dependent and p53-independent apoptosis can be mediated by miR-34a[117]. Nevertheless, some evidence points to p53's involvement in the development of NAFLD and NASH; however, more research is needed to show if p53 actually have a critical role in the development of NAFLD-mediated HCC[86].
Iron overload
Iron is a crucial mineral that is involved in several metabolic processes, including heme production, the formation of the Fe-S cluster and the transportation of oxygen by haemoglobin[118].
Nevertheless, there can be serious implications when iron homeostasis is disturbed, whether as a result of hereditary or environmental factors, such as hepatic fibrosis, cardiomyopathy and arthropathy,endocrine abnormalities[119,120]. Crucially, having too much iron raises the risk of developing a variety of neoplasias, such as breast cancer, colorectal cancer and HCC[121]. Dietary iron excess, primarily in beer cooked in iron pots, is closely linked to a higher risk of HCC in several areas of sub-Saharan Africa[122]. HCC has been demonstrated experimentally in Wistar rats on iron-rich diet[86,123]. Multiple rodent models and investigations of human HCCs have connected higher iron content with increment in 8-oxo-2-deoxyguanosine adducts and oxidizing agents like malondialdehyde, suggesting that ROSinducing DNA alterations are one pathway involved in iron overload-linked HCC genesis[86,124].
Moreover, there is proof that an excess of iron directly affects p53 activity[86]. A high-iron diet causes p53 protein levels in the liver of C57BL/6 mice to drop[125]. Additionally, male Sprague-Dawley rats exhibit a rise in MDM2 and a reduction in p53 in the liver when fed a high-iron diet over an extended length of time[126]. Another chemical mechanism for why there are fewer p53 molecules in the blood when there is too much iron is that p53 is linked to heme, transferred to the cytoplasm and then destroyed by the proteasomal pathway in HepG2 cells[125]. As a result, p53 levels are decreased by both iron overload and dysregulated heme, which aids in the development of HCC[86]. It's interesting to note that p53 also affects intracellular iron contentviatransactivating iron-sulfur cluster enzyme 2,which results in less efficient iron uptake[127,128]. As a result of chronic iron overload, decreased p53 function raises intracellular iron levels, which further encourages the development of HCC. It is important to emphasized that people with hereditary hemochromatosis have greater frequencies of p53 mutations (64%-71%) than those with sporadic HCC, indicating a possible involvement of p53 in the development of HCC brought on by iron overload[129,130]. 45% A>C transitions and 33% G>C transversions, involving two hotspots at codons 275 and 298, are seen in HCC tissues from hereditary hemochromatosis[86,130]. The p53 mutation spectrum in the research utilizing British families with hereditary hemochromatosis is made up of 60% A>G transitions and 40% A>T transversions, though[129]. However, it is still unclear if iron excess does in fact cause HCC in animal models in a p53-dependent manner[86].
HBV
According to estimates, 248 million people worldwide have chronic HBV infection and are hepatitis surface antigen positive[131]. HBV is the main contributor of HCC, with chronic HBV infection accounting for the majority of cases[86,132]. Repeated episodes of immune-linked hepatocyte death and consequent tissue regeneration can result in cirrhosis of the liver and HBV-mediated HCC carcinogenesis[133]. Importantly, 10%-30% of HCCs linked to HBV do not develop in the context of cirrhosis,pointing to additional oncogenic pathways involved in the development of HCCs linked to HBV[86,134].
The genome of HBV, a circular, partly double-stranded DNA virus, has four overlapping open reading frames: A core area, a surface region, a polymerase region and an X region[86]. These four regions together develop seven viral proteins: Precore, polymerase, S, M, core, HBx and L[135,136]. The HBx protein, which is crucial for viral replication and is most connected to the development of HCC, is one of these[136]. In fact, HBx causes HCC in transgenic mice models by confining p53 to the cytoplasm[137,138].
Additionally connected to hepatocyte apoptosis is HBx[86]. In many situations, HBx prevents apoptosis by adhering to and sequestering p53 in the cytoplasm in addition to raising levels of the antiapoptotic protein surviving[69,139]. Additionally, HBx is said to suppress tumour growth factor (TGF)-mediated apoptosis[140]. On the other hand, in rare circumstances, HBx has been demonstrated to cause apoptosis without the involvement of p53[141,142]. The double functions of HBx in hepatocyte apoptosis and its connection to the development of HCC, therefore, call for additional research[86].
Although there is no concrete proof for it, HBx variants with C-terminal truncations (Ct-HBx) are typically found in HCC and may potentially have a role in the progression of HCC[143,144]. In numerous cell lines, Ct-HBx suppresses apoptosis and encourages hepatocyte growth[145]. It has also been demonstrated that Ct-transcriptional HBx's repression of USP16 (ubiquitin-specific peptidase 16)enhances the tumourigenicity and stem-like characteristics of HCC cells[86]. Additionally, like HBx, Ct-HBx links to p53 and blocks p53-linked apoptosis[135]. Likewise, few Ct-HBx variations have the capacity to inhibit GAS2, a modulator of p53-linked apoptosis, from expressing its mRNA[146]. As a result, USP16 is downregulated and p53-mediated apoptosis is inhibited, which may help explain how Ct-HBx contributes to the pathogenesis of HBV-related HCC[86]. Given that p53 mutations are uncommon in HBV-related HCC, are linked to advanced disease and are inhibited by both HBx and Ct-HBx, suppression of p53 activity might be more advantageous for HBV-linked HCC carcinogenesis than p53 mutation[147]. Crucially, HCC patients with wild-type p53 outlive those with p53 mutations in terms of both overall survival and recurrence-free survival[86].
HCV
According to estimates, 184 million people worldwide have anti-HCV positivity and those who have HCV have a 15-to-20-fold higher risk of developing HCC[148]. HCV is a single open reading, positive sense, 9600 nucleotide long single-stranded RNA virus[149]. The HCV genome produces a polyprotein that is broken into nine viral proteins, encompassing structural proteins (C, E1 and E2) and nonstructural proteins (N, S and S2) (p7, NS2, NS3, NS4A, NS5A, NS5B)[86,150]. Although cirrhosis is the primary setting for the great majority of HCCs connected to HCV, there is some proof that the HCV viral proteins have the capacity to cause cancer[134]. Particularly, both p53-dependent and -independent mechanisms have been proposed to link the development of HCC to the HCV core, NS3 and NS5 proteins[86].
Without the presence of cirrhosis, HCC definitely instantaneously arise in transgenic mice that produce the HCV core protein[151]. The Wnt-β-catenin pathway is upregulated, ROS levels are increased and Fas- and tumor necrosis factor (TNF)-mediated apoptosis is inhibited by HCV core protein[86]. The core protein alters the subcellular localization of p53 to the perinuclear area and nuclear granular structures, as well as its post-translational changes, such as phosphorylation and acetylation, in HeLa and HepG2 cell lines[86]. This is significant because it reduces p53 activity[152]. Additionally, the core protein activates SIRT1, which deacetylates p53, impairing p53-dependent apoptosis in HepG2 cells. Thus, both p53-dependent and -independent mechanisms by which HCC is caused by HCV core protein are likely[86].
Another HCV protein that could alter human hepatocytes is called NS3, a non-structural HCV protein that increases cyclooxygenase-2 and activates mitogen-stimulated protein kinase[153]. In HeLa and NIH3T3 cells, NS3 also forms complexes with p53[154] and inhibits p53's transcriptional activity in NIH3T3 and Huh7 cells. Furthermore, overexpressing NS3 can cause NIH3T3 cells to convert and cause tumours in animals[155]. It is yet unknown, nevertheless, whether or not p53 is required for NS3-mediated transformation. In transgenic mice models, the non-structural HCV protein NS5A can result in steatosis and HCC[86]. It has been demonstrated that NS5A inhibits Bax-linked apoptosis, transactivates c-fos and prevents TNF-linked apoptosis without the need for p53[156]. However, p53's transcriptional activity can be inhibited when NS5A binds to and colocalizes with p53 in the perinuclear membrane.Additionally, NS5A interacts with hTAFII32 at the nucleoplasm membrane and prevents it from stabilizing p53, which prevents p53-mediated apoptosis in Hep3B cells[157]. As a result, NS5A promotes the growth and progression of HCC through both p53-dependent and -independent mechanisms[86].
Relationship between HCV Core protein and p53 in HCC
The structural viral protein known as the HCV core protein encases the viral genomic RNA. According to its subcellular localization and the subsequent impact on the cell cycle inhibitor p21, the core protein may also have contradictory effects on cell growth, supporting both apoptosis and cell proliferation[158,159].
The innate version of the HCV core protein is synthesized first (amino acids 1–191) and a mature form (amino acids 1–173) is created during processing[61]. The innate form and the mature form, form a heteromultimer in the cytoplasm, preventing the mature form from being transported to the nucleus.The innate form activates p53 in the cytoplasm and p53 then increases the production of p21 by acting as a transcription factor for p21. It is unclear how the innate form stimulates p53, but because it binds to the protein, it may do so by stabilizing the protein[159,160]. p21 expression can be decreased by a process unrelated to p53 if the amount of mature core protein penetrates the nucleus and surpasses the innate form's binding capacity[161,162]. A core-responsive region in the p21 promoter precisely overlaps a butyrate/TGF-RE. In this instance, core protein stimulates a butyrate route to activate p21viathe element[73].
Additionally, the HCV core protein controls p73, a tumour suppressor protein belonging to the p53 family[61]. In addition to being engaged in neurogenesis and innate immune responses, p73 also appears to have a significant role in the development and maintenance of malignancies[163]. The HCV core protein is translocated into the nucleus as a result of its interaction with p73. Additionally, in a p53-dependent way, the association with the core protein blocks p73-α, but not p73-β, dependent cell growth arrest. Consequently, the impact of the HCV core protein on p73 function may play a role in HCV pathogenesis[61].
Relationship between HCV NS3 protein and p53 in HCC
The HCV NS3 protein may cause host cells to become hepatocarcinomas at an early stage. Additionally,it has been suggested that it might result in p53 gene mutation and hepatocyte transformation, however this is debatable[164,165]. NS3 and wild-type p53 combine to form a complex[164]. The oligomerization domain, located close to the C-terminus of wild-type p53[166] (amino acids 301-360), is crucial for the formation of this combination with NS3. In line with this discovery, NS3 protein can specifically and dose-dependently suppress the promoter activity of p21viaaltering the activity of p53[61]. The impact works in concert with that of the HCV core protein and is not cell-type specific.
Relationship between HCV NS5A protein and p53 in HCC
A phosphoprotein between 56 and 58 kDa, the HCV NS5A protein. Although NS5A is a member of the viral replicase complex, which is located on the cytoplasmic side of the endoplasmic reticulum, along with other virus-encoded proteins, its function in viral replication is yet unknown[61]. Phosphorylation and possible proteolytic processing of NS5A into lower molecular weight forms that can translocate to the nucleus are examples of post-translational changes[167]. Truncated forms of NS5A can act as transcriptional activators, but other now has discovered interrelations of NS5A with cellular proteins,such as those of the transcription activation machinery, cellular signalling apparatus and cell-cycleregulatory kinases, suggesting that it plays a pleiotropic role in HCV-host interrelations[167,168].Numerous of these interactions prevent the apoptotic cellular response to persistent HCV infection, that raises the possibility that NS5A plays a role in the development of chronic liver conditions and HCC linked to HCV infection[61].
By locating p53 in the perinuclear area, NS5A can prevent it from binding to certain DNA sequences.Inhibiting p21 expression and p21-mediated apoptosis in this manner[169]. Additionally, p53 and TATA-box-binding protein (TBP) connect to NS5A, generating a heteromeric complex that prevents p53 and TBP from attaching to their respective DNA binding regions. In addition, p53-TBP-excision repair cross-complementing factor 3 complex formation is inhibited by NS5A[61]. Additionally, the NS5A protein may prevent p53 from activating by securing the crucial coactivators of p53, hTAF(II)32 and hTAF(II)28. Recently, it was discovered that the protein kinase PKR, which is activated by interferon(IFN), is inhibited by NS5A. Thus, NS5A may be involved in HCC's IFN resistance[169,170].
OTHER MECHANISMS OF PROGRESSION TO CIRRHOSIS AND HCC
Other potential risk factors for the development of HCC include genetic predisposition, prenatal anomalies, chemical exposures (aflatoxin or food tainted with arsenic), and autoimmune illnesses of the body’s liver[106].
Patients with a number of congenital defects are more likely to develop liver cirrhosis and HCC.These consist of hemochromatosis, alpha-1-antitrypsin deficiency, Wilson's disease, and hereditary tyrosinemia[171]. AFB1-induced HCC is caused by a number of processes, some of which involve the development of mutagenic and carcinogenic intermediates and adducts. Food contaminated by the fungusAspergillus flavusandAspergillus parasiticusreleases aflatoxins. There are several chemical changes that happen as a result[106].
AFB1 is transformed into the following known mutagenic or carcinogenic compounds: 8,9-dihydroxy-8-(N7), aflatoxin-B1, aflatoxin B1-8,9 exo-epoxide. Aflatoxin B1 form aminopyrimidine adduct guanyl-9-hydroxy aflatoxin B1 adduct[106]. Additionally, these intermediates and adducts have the ability to directly cause a mutation at codon 249 of the p53 tumour suppressor gene[106]. This changes the amino acid arginine to serine, reversing the gene's ability to prevent tumour growth[106].According to certain reports, AFB1 and HBV may work together to cause HCC[172]. Additionally,additive interactions have been documented. In a systematic review, Ogunwobiet al[106] showed a connection between autoimmune hepatitis (AIH) and an elevated risk of developing HCC in individuals with liver cirrhosis. The chance of developing liver cirrhosis from AIH was determined to be less than the probability of developing the condition as a result of HBV, HCV, or primary biliary cholangitis. But the risk of HCC and liver cirrhosis from AIH is clinically considerable[106].
MDM2-P53 INTERACTIONS IN HCC
It has been demonstrated that MDM2 inhibits p53 gene transcription and translation in addition to adversely controlling p53 functions through ubiquitination and degradation[173]. The cell fate as well as the balance between MDM2 and p53 may be impacted by factors that control the ubiquitination of MDM2 and p53[174]. The connections between MDM2 and p53 are regulated by various mechanisms[14,175]. Ones of these interactions could be.
RPs
RPs have a substantial impact on nuclear strain, cell cycle arrest and apoptosisviaactivating the MDM2-p53 regulatory loop[176]. Knockdown of nuclear proteins can cause nuclear/ribosomal stress by sequestering MDM2 and inhibiting its ubiquitin ligase function against p53[177], which in turn causes nuclear/ribosomal stress[35]. It has been demonstrated that ribosome biogenesis interruption leads to p53 stability. This is caused by a number of RPs that are linked to MDM2 and restrict its action for p53 being released from the nucleolus[35]. As a result of blocking E3 ubiquitin ligase, the subunit RPs rpL5,rpL23 and rpL11 have demonstrated the capacity to attach to MDM2. According to Zhanget al[178], the human protein HDM2, which is the MDM2 ortholog, interacts to RP L11[14]. The docking site is located in the middle of HDM2, which prevents HDM2-associated p53 ubiquitination and destruction and restores p53-linked transactivation and p21 protein deposition. The research confirmed the idea that L11 controls HDM2 negatively[14].
Ribosomal RNA
The most prevalent element found in ribosomes is ribosomal RNA, which is the RNA portion of the ribosome and is necessary for protein synthesis[14]. In human tumour cells, it has been discovered that RNA polymerase I subunit A (POLR1A) gene silencing results in a decrease of rRNA production that stabilizes p53 without altering the integrity of the nucleolus. The suppression of the MDM2-mediated p53 breakdown was what caused p53 to stabilize. However, rRNA production down-regulation that was accompanied by a decrease in protein synthesis did not result in the stability of p53[14]. The results showed that p53 functions in mammalian cells are controlled by the equilibrium of RP and rRNA production[36].
MicroRNAs
Small non-coding RNAs called microRNAs (miRs) regulate the transcription and post-transcriptional expression of many target genes. In cancer, their action is typically dysregulated[14]. Micro-RNAs generally decrease in human malignancies, suggesting that they could be involved in tumour suppression. Therefore, by focusing on oncogenes or tumour-suppressor genes, miRs can perform a variety of biological roles[14]. The first miR-34 family members to be implicated in the p53 tumoursuppressing network was identified in 2007 and are known as miR-34a, miR-34c and miR-34b[179]. The expression of members of the microRNA family (miR-34a-c) evidenced the p53 condition and the genes encoding miR-34 family members were transcriptional domains of p53in-vivoandin-vitro, according to the authors' comparison of the microRNA expression profiles of cells with and without p-53 deficiency[14]. As a result, the p53 network inhibited the development of tumoursviaa variety of co-ordinated interactions and multiple transcriptional targets, such as the function of the miR-34 family members in preventing uncontrolled cell growth[14].
Biet al[180] investigated the function of miR-125a in HCC. MiR-125a expression was found to be downregulated in HCC tissues and cell lines and it was linked to aggressive pathological characteristics[14]. According to the research, miR-125a targets vascular endothelial growth factor A and matrix metalloproteinase II, which both functionin-vivoandin-vitroto prevent the progression and metastasis of HCC. As a result, miR-125a overexpression might be an effective treatment for HCC.
Experiments on miR-125b's function revealed that it negatively regulated p53 and p53-caused apoptosis in cells under stress, implying that it had a tumourigenic effect, for example[181]. miR-122,one of the several miRs found in liver tissue, has been demonstrated to function as a tumour suppressor in HCC[14]. Poorly differentiated cancer, big tumours, metastasis and a poor prognosis are usually linked to decreased miR-122 expression[182]. Simerzin and colleagues recently demonstrated that miR-122*, the passenger strand of miR-122, targeted MDM2 and had an active role in the association between MDM2 and p53. The levels of miR-122* and MDM2 were significantly inversely correlated in human HCC samples. By highlighting the tumour-suppressor properties of miR,in-vivoresearch demonstrated that miR-122* was capable of preventing the formation of tumours[14]. Inhibiting miR-122 using an antagomiR-122 (miR inhibitor) caused miR-122* to build up, MDM2 to be suppressed and the p53 protein to be increased. The research revealed that miR-122*, the miR-122 passenger strand,controlled the function of p53 by concentrating on MDM2. As a result, miR-122* exhibits tumoursuppressive action that was previously only associated with miR-122. Additional research is required to determine whether it is useful as a treatment for HCC as well as what key distinctions occur among miR-122* and miR-122[14].
Kimet al[183] discovered that p53 increased the expression of members of the miR-200 and miR-192 families and explained the function of p53 in controlling the EMT in HCCviathe development of particular effector miRs, such as miR-141, miR-192, miR-194, miR-200b, miR-200c and miR-215[14].These miRs specifically targeted the zinc-finger E-box-binding homebox 1 and 2 (ZEB1 and ZEB2) and EMT-activity transcription factor 2.ZIB1andZEB2genes were repressed in human HCC and HCC cell lines by p53 overexpressed miRs. This reduced EMT resulted in enhanced E-cadherin and reduced vimentin expression[14].
LIM Domain protein enigma
It has been demonstrated that the protein Enigma, PDLIM 7 (Enigma LIM), which has a LIM domain,interacts with proteins included in protein kinases and that the Enigma gene has been detected in stomach and liver malignancies in humans[14]. It's also been demonstrated that evenin-vitro, in human hepatoma cell lines and in mice embryonic fibroblasts, the LIM domain protein Enigma actively links with MDM2 to create a complex with p53[14]. Furthermore, it has been demonstrated that the Enigma caused p53 breakdown by reducing MDM2's ability to ubiquitinate itself and boosting its ability to bind to p53[40].
SRF
Serum growth factors were capable of rapidly stimulating the expression of HDM2 in a well-studied non-transformed human fibroblast model, as demonstrated by Phillipset al[184]. The authors noted that the increased HDM2 protein, that also accompanied growth factor activation, was mainly the consequence of phosphatidylinositol-3 kinase-dependent stabilization of the HDM2 protein along with a global rise in protein synthesis in their investigation, which investigated the mechanistic reaction for these variations[14]. According to a new study, SRF induction enhanced Enigma transcription,stabilizing HDM2, which then caused p53 to degrade[14]. The authors saw these modifications in the liver given the hepatocyte growth factor. Additionally, they discovered that MDM2 co-expressed with SRF and Enigma, but not p53, in the human stomach and liver tumours. Enigma was also discovered to decrease p53-linked apoptosis in cell lines and a mouse xenograft model, which boosted cell viability and produced chemoresistance[14].
PRL-1
Several malignancies, including colorectal[185], gastric and liver tumours, and especially metastatic cancers, have been discovered to overexpress the protein tyrosine phosphatase family of the PRL family.PRL-1, PRL-2 and PRL-3 displayed a higher level of structural sequence similarity within this family.According to Minet al[186], overexpression of PRL-1 prevented p-53-mediated apoptosis and decreased levels of endogenous and exogenous p53[14]. The ubiquitination of p53 and subsequent proteasomal degradation were the mechanisms by which p53 was down-regulated. The stimulation of PIRH2 (p53-induced RING-H2) transcription and the promotion of MDM2 phosphorylationviaAkt signalling were the two main strategies used to ubiquitinate PRL-1[14]. These results suggested that the growth of cancer was aided by the down-regulation of p53 caused by a PRL-1-induced negative feedback mechanism. Additional research by the same team showed that PRL-3 activated PIRH2 and MDM2 in cancer cells, negatively regulating p53 in a manner akin to PRL-1[187].
MDM2 binding protein
The MDM2 binding protein (MTBP) was demonstrated to have metastasis-suppressing properties in earlier investigations. Additionally, it has also been proven that MTBP prevented infiltration irrespective of p53 status[172]. Currently, Biet al[188] established the growth inhibitory action of MTBP in HCC and observed that, when compared to nearby non-tumour tissue, MTBP expression was reduced in almost 70% of human HCC cells[14]. The local invasion of lymph node metastases was also linked to this decrease. Regardless of the p53 status, the transcription rates of MTBP were found to be adversely linked with the ability of HCC cells to migrate[14]. These findings supported the findings of Vlatkovićet al[189], who showed that low MTBP was linked to a poor prognosis for head and neck malignancies. Moreover, there are currently a number of holes in this field. For instance, it is yet unclear how HCC cells control the expression of MTBP. Additionally, it is unclear how functionally related MTBP and MDM2 are. We have to understand why p53 is not required for these modifications in the interim[14]. The existence of a single nucleotide polymorphism (SNP309) in the MDM2 promoter is another element that might influence how p53 interacts with MDM2. It has been demonstrated that this factor raises the affinity of the transcriptional activator Sp1, leading to greater levels of MDM2 RNA and protein as well as a resulting attenuation of the p53 pathway[14]. The authors' suggestion that SNP309 serves as a rate-limiting event in carcinogenesis stems from studies showing that such mechanisms enhance the development of tumours in humans[190].
MECHANISMS OF P53 THERAPY FOR HCC
HCC typically harbours p53 mutations[191]. Despite carrying wt-p53, the majority of HCCs have abnormalities in the p53-mediated apoptotic pathway[192]. In HCC, p53 might occasionally be wildtype but inactive[193]. The expression of wt-p53 can be induced by signals to and activation of p53,which suppresses the altered phenotype of hepatocytes and boosts the actions of radiation therapy and chemotherapy[194]. In HCC, p53 can be partially or completely repressed and single tiny compounds,like the well-known antimalarial medication quinacrine, can activate p53[195]. Exogenous wt-p53 can be delivered into cancer cells by gene delivery, which effectively slows the growth of tumours that contain both mutant and wild-type p53[196]. The oncogene MDM2 generates a phosphoprotein that builds a strong complex with mutant as well as wild-type p53 protein, blocking p53-mediated transactivation and causing p53 breakdown[197]. MDM2 also has a p53-DNA binding site[195]. The MDM2-p53 autoregulatory feedback loop[198] controls the transcription of the MDM2 gene and the activity of the p53 protein. The amount of wild-type p53 protein can control the expression of the MDM2 gene and the MDM2 protein can interact with p53 to reduce its capacity to function as a favorable transcription factor at the MDM2 gene-responsive region[195]. To boost functional p53 levels and p53-mediated therapeutic efficacy, several methods have now been used to disrupt MDM2-p53 interaction[199]. The proteinsecreting pathway is significantly regulated by Adenosine diphosphate (ADP)-ribosylation factor (ARF)proteins[200]. ADP and adenosine triphosphate are converted to adenosine in a human HCC cell line and treating HCC cells with adenosine can decrease their development and activate caspase-3, showing that adenosine is a cytotoxic apoptotic agent[195]. ARF, which is regarded as a tumour suppressor[201],can start the cellular response to abnormal oncogene stimulation by attaching to and preventing MDM2 from functioning[195]. Like the murine protein, the human protein p53's capacity for transactivation can be inhibited by HDM2, which is MDM2's human counterpart[202]. Different locations on MDM2 are where ARF and p53 bind. According to MDM2's mediation, the ARF-p53 complex is created, which can improve p53 stability[203]. Successfully inducing p53-triggered tumour cell death can be achieved by blocking the routes for p53 breakdown, either through over-expression of ARF or by disrupting the connection between MDM2 and p53[195].
High levels of exogenous p53 protein might have therapeutic benefits for HCC in two ways: (1) By increasing tumour cell apoptosis by preventing cell proliferationviaa number of physiological mechanisms and[123] by making HCC more susceptible to certain chemotherapeutic drugs[195]; and (2)Additionally, due to the wt-p53 protein's angiogenesis blocking properties, p53 reduces the expression of genes related to angiogenesis[204].
Due to medication resistance, treating HCC is extremely challenging. Reversing multiple drug resistance in human HCC cells and figuring out the underlying processes are both important[195]. In HCC cells expressing mt-p53, P-glycoprotein expression and function are increased, as are the products of numerous drug resistance genes (MDR)[205]. Expression of mt-p53 decreases the absorption of chemotherapeutic drugs by HCC cells and increases their treatment resistance. MDR transcription is inhibited by wt-p53 as opposed to mt-p53, which increases MDR expression[206]. Due to the reduced expression of P-glycoprotein encoded by MDR, HCC cells become more sensitive to chemotherapeutic drugs when wt-p53 function is restored in these cells[195].
THERAPEUTICS PRODUCTS WITH P53 STATUS AS A TARGET
Currently, the most frequently usedin-vitroandin-vivotherapeutic agents for the treatment of HCC are the oncolytic virus and recombinant adenovirus p53 (rAd-p53)[195]. Both the expression of p53 and p21 proteins is observed to be up-regulated in cells following the introduction of exogenous wt-p53 generated by the recombinant adenoviral vector. It is possible to stop cell division and apoptosis.Oncolytic viruses are a group of flawed viruses[207] that cannot reproduce in healthy cells but can expand in tumour cells, ultimately lysing them[195].
Recombinant adenovirus p53
Human adenovirus vector processes are typically utilized for the expression of proteins in humans or other species with a few benefits due to their wider host range and lesser pathogenicity to humans, have a binding attraction for epithelium[208], which is essential as the majority of human tumours are of epithelial origin and serotype 4 of species E[209] have a specific binding affinity for HCC cells[208] . It has been determined that the E1 region of the adenovirus, a subregion of the viral genome seen in converted cells, is what causes transformation[195]. Exogenous DNA is used to substitute the E1 region in the construction of recombinant human adenoviruses, which results in replication defects and a negligibly high level of acute toxicity[210]. Recombinant adenoviral vectors that express wt-p53 (Adp53) allow for the transfection of the p53 gene into HCC cell lines[195].In-vivotumour growth of neighbouring non-transduced cells can be inhibited by tumour cells transduced with the wt-p53 gene,according to experiments. This suggests that Ad-p53 is also anti-angiogenic[195,211], at least in part because of the bystander effect caused by the transfer of the wt-p53 gene on neighbouring tumour cells.
Oncolytic viruses
Oncolytic viruses that interfere with the key surveillance pathways, as those mediated by p53, might multiply selectively in them and cause lysis since HCC cells with p53 abnormalities have abandoned their cellular surveillance systems[195]. Adenoviruses have E1A and E1B genes, with E1A being an apoptosis-inducing gene and E1B being an apoptosis-inhibiting gene[195]. The p53 gene is bound to and rendered inactive by the 55kDa protein from the E1B-region of the adenovirus[212]. The 55-kDa E1B protein is not produced due to a loss in E1B and the mutant adenovirus known as ONYX-015[213] can only multiply in cells lacking wt-p53. The E1B55K-defective adenovirus ONYX-015 is a prototype for oncolytic viruses that may specifically reproduce in and kill p53-deficient HCC cells[195]. Unless cancer gene therapy is properly planned based on the biology of a particular tumour type[214], it is unlikely to be successful. Another E1B 55kDa-deficient adenovirus with a mouse endostatin gene has been developed for antitumour activities against HCC and is known as CNHK200-mE to increase the effectiveness of such oncolytic viruses[195]. A novel strategy known as a gene-viral vector or gene-viral therapeutic system has been developed using the synergic activity of a carrier virus and therapeutic gene[195]. In this method, an anti-tumour gene is inserted into the genome of a replicative virus specific for tumour cells in order to merge the benefits of gene and virus therapies. CNHK300-murine endostatin (CNHK300-mE) is created using the human telomerase reverse transcriptase promoter to activate the production of the adenovirus E1A gene and clone the therapeutic gene mouse endostatin into the adenovirus genome, indicating promising results in the treatment of HCC[195] (Table 1).
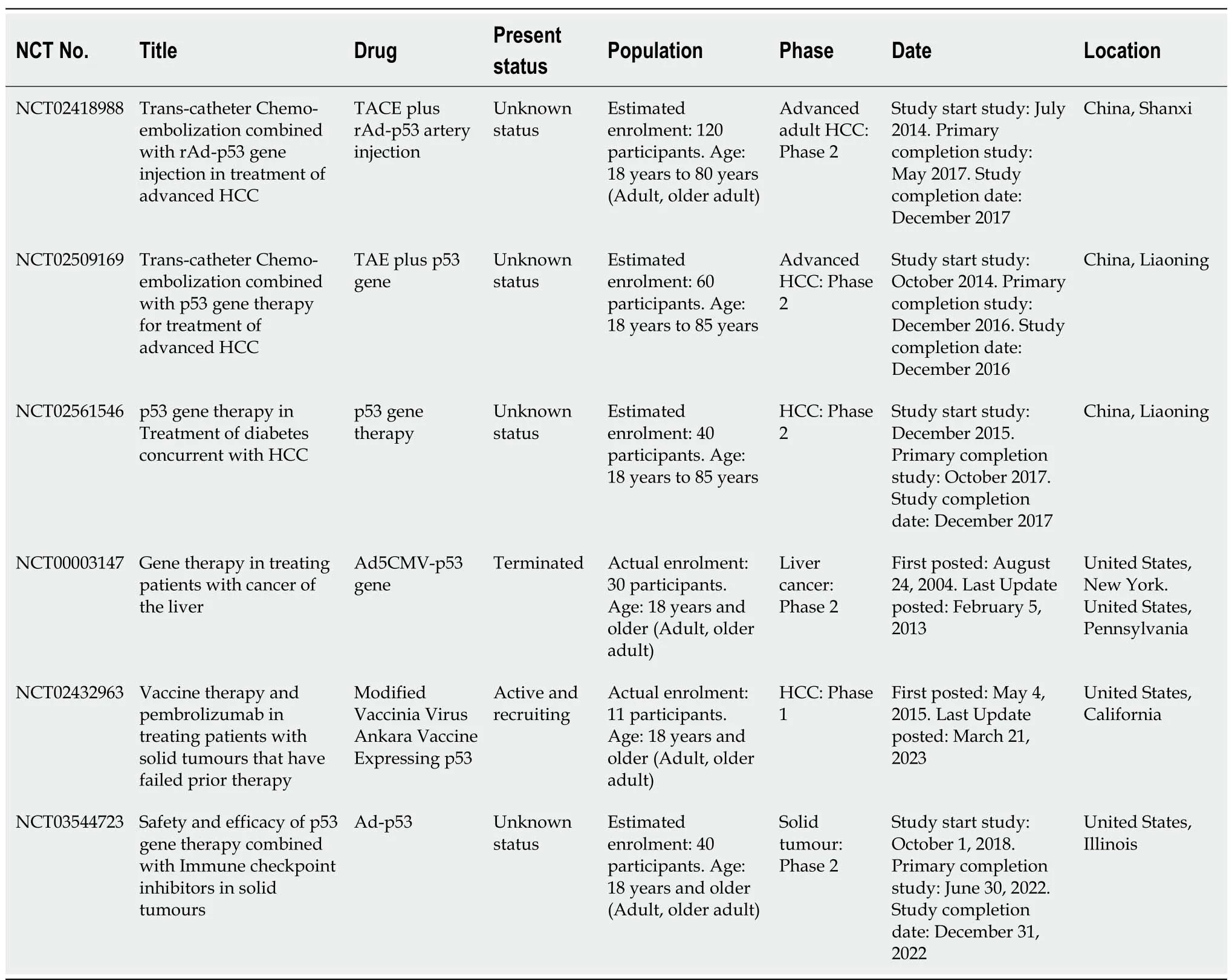
Table 1 List of clinical trials undergoing for hepatocellular carcinoma targeting p53
P53 GENE IN CHEMORESISTANCE IN HCC
Through the direct inhibition of apoptotic pathways, the buildup of mutant p53 with gain of function can cause tumours to become resistant to treatment[215]. p53 has been linked to chemotherapeutic treatment resistance in HCC. Cisplatin, also known as cis-diamminedichloroplatin (CDDP), is an essential part of standard treatment plans for tumours of the gastrointestinal, pulmonary and genitourinary systems[216]. The response rate to CDDP in systemic monotherapy for HCC is 15%[217],however multidrug regimens containing this medication produce higher response rates[218]. Because arterial infusion schemes with CDDP reach larger concentrations of the drug inside the tumour and consequently exhibit a more potent anticancer effect, response rates range from 41% to 61%[219]. In most HCC cell lines, regardless of p53 status, a prior study revealed that CDDP has a synergistic effect with tumour necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis[218]. But an in-depth examination of the Bel7402 (p53 wild type), Huh 7 (p53 mutant) and Hep3B (p53 deficient) cell lines revealed that the p53 status is only partially necessary for CDDP to reverse TRAIL resistance in HCC cells[220]. Thus, p53 is linked to the effectiveness of CDDP in inducing apoptosis. Later research revealed that defective p53 contributes to CDDP resistance[218]. In HepG2 cells, the long interspersed nuclear element-1 ORF-1 protein (LINE-1 ORF-1p) enhances CDDP and epirubicin resistance by suppressing the production of the p53 gene[221]. As a result, HCC cells are more likely to be CDDP resistant when p53 expression is downregulated[218].
The cornerstone of chemotherapy for HCC is doxorubicin (Adriamycin, ADA), but doxorubicin resistance prevents these patients from receiving a complete course of treatment. Doxorubicin activates the p53 pathway to cause apoptosis in human HCC cells[222]. In contrast to wild-type p53, which enhances doxorubicin chemosensitivity in the drug-resistant human HCC cell line Bel7402/5-fluorouracil (5-FU), mutant p53 enhances doxorubicin resistance in HCC cell lines[205]. Doxorubicin sensitivity is reduced by p53 activation inhibition[223]. N-myc downstream regulated gene-1, a downstream target of p53, also contributes to doxorubicin resistance in HCC cells[224]. 5-FU and vincristine resistance in HCC cells is linked to p53 mutation and inactivation addition to doxorubicin resistance[218].
For HCC patients, doxorubicin and sorafenib combined therapy produce better results than doxorubicin alone does in terms of median time to progression, overall survival and progression-free survival[103]. These findings suggest that the use of sorafenib in combination with doxorubicin is effective in the treatment of HCC[218]. By reducing Bcl-2 expression, microRNA-34a, a specific target of p53, improves the susceptibility of human HCC cells to the anticancer impact of sorafenib, amplifying sorafenib-induced apoptosis and damage[225]. CDDP, doxorubicin, 5-FU and other treatments are ineffective against HCC cells due to p53 mutation and inactivation, among other factors. The G245D mutation in p53 reduces the chemosensitivity of HCC cells by making them resistant to histone deacetylase inhibitors[218]. Contrarily, dominant-negative p73 (Delta Np73) can suppress p53 function,leading to treatment resistance. Chemoresistance is caused by deltaNp63alpha's direct interference with the p53 family target genes' ability to activate transcription[226]. The mechanism underpinning p53-related chemoresistance may be started or controlled by both extrinsic and intrinsic factors because chemotherapy-induced apoptosis involving p53 in HCC is mediated by extrinsic and intrinsic pathways[218].
p-53 repression leads to doxorubicin resistance in HCC
Dox is the cornerstone of chemotherapy for HCC, but Dox resistance prevents patients with HCC from receiving an effective course of treatment[227]. Dox activates the p53 pathway in human HCC cells to cause apoptosis. Maet al[227] looked at the connection between Lgr5 and p53 expression in HCC in order to clarify the molecular mechanism of Dox resistance[227]. They discovered that Lgr5 stimulates p53 degradation in HCC cell lines and that there is an adverse connection between Lgr5 and p53 expression in HCC tissues. Target gene PDCD5 was found; PDCD5 is a protein involved in apoptosis and is thought to act as a tumour suppressor in a variety of tumour types, particularly lung cancer,breast cancer, and glioma[228,229]. In the presence of DNA damage, PDCD5 can interact with and stabilize p53 to encourage apoptosis[230]. They observed that PDCD5 serves as an independent predictive factor and serves as an antioncogene. Drug resistance caused by Lgr5 is only partially reversed by PDCD5 overexpression. They initially created transfected HCC cell lines that overexpressed Lgr5 and PDCD5, and we discovered that neither protein could enhance the expression of the other at the transcriptional and posttranscriptional stages[227]. This information allowed us to expand our understanding of the interaction between Lgr5 and PDCD5. In addition, they discovered that Lgr5 maybind with PDCD5 in the cytoplasm and stop it from translocating. As a result, PDCD5 was unable to bind to p53 and translocate to the nucleus, stabilizing p53. This suggests that Lgr5 Lowers the effective level of PDCD5 as opposed to the overall concentration of this protein. In HCC cells treated with Dox or even untreated cells, high levels of Lgr5 could lower p53 protein levels and prevent apoptosis[227].They overexpressed PDCD5 in HCC cells that had been stably transfected with Lgr5 to examine how Lgr5 affected PDCD5. They discovered that this decreased the cells' ability to resist Dox and caused them to undergo apoptosis, which suggests that PDCD5 overexpression is antagonistic to Lgr5. These observations indicated that Lgr5 could prevent PDCD5 from translocating into the nucleus by binding directly to PDCD5 with its N-terminal extracellular domain and causing p53 to degrade. Recent studies demonstrating that PDCD5 may move from the cytoplasm to the nucleus after Dox treatment[231]reinforce this conclusion. Additionally, we discovered that Lgr5 prevents Dox-induced apoptosis in HCC cellsviainhibiting the p53 pathway[227].
They offer a hypothesis for a brand-new mechanism of Dox resistance in HCC cells, one that emphasizes the crucial function of Lgr5, which is effectively triggered by this chemotherapy medication.When the N-terminal extracellular domain of the triggered Lgr5 directly binds to PDCD5, it inhibits PDCD5's nuclear translocation[227]. This is how the induced Lgr5 functions as a direct negative regulator of PDCD5. The lack of p53 stabilization caused by the decreasing PDCD5 levels within the nucleus causes an EMT-like phenotype, which inhibits apoptosis and eventually results in Dox resistance[227].
CONCLUSION
The recent literature was examined in this review to show the negative impacts of MDM2-p53 axis malfunction on the transformation of hepatoma cells. Despite numerous studies proposing tumour cellspecific signaling pathways that affect the MDM2-p53 axis, the mechanisms causing malfunction in this axis during the progression of HCC are intricate. To better diagnose and treat HCC at an early stage,future studies should concentrate on the characteristic features of MDM2-p53 axis malfunction in chronic progressive liver diseases that are closely linked to the onset and advancement of HCC. The balance of the MDM2-p53 axis in chronic liver illnesses could be modulated by a number of molecules,and it has been discovered that HCC tissues have severely altered levels of these molecules. By emphasizing on the malfunction of the MDM2-p53 axis during the conversion of healthy hepatocytes into hepatoma cells, we intended to offer some guidance for techniques to cure pre-HCC.
ACKNOWLEDGEMENTS
We would like to thank Dr. A. P. Pawar, Principal, Poona College of Pharmacy, Bharati Vidyapeeth(Deemed to be University), for providing the necessary facilities to carry out this research work.
FOOTNOTES
Author contributions:Choudhary HB, Mandlik SK and Mandlik DS were responsible for literature research and drafting of a manuscript; All authors have read and approved the manuscript.
Conflict-of-interest statement:No conflict of interest in among the authors.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:India
ORCID number:Deepa S Mandlik 0000-0001-8505-7038.
S-Editor:Fan JR
L-Editor:A
P-Editor:Fan JR
