潜在马铃薯晚疫病抗性基因的表达和SNP标记分析
2023-06-11李晓川王朝海周平马维陈军陆燚吴显王宗明吴瑞宋治豪马杰付毅
李晓川 王朝海 周平 马维 陈军 陆燚 吴显 王宗明 吴瑞 宋治豪 马杰 付毅


摘要 从马铃薯参考基因组中筛选出了433个NBS-LRR类基因,并绘制这些基因在基因组中的分布图,这些基因的其中一部分成簇分布。马铃薯基因组测序协会(PGSC)的转录组数据分析显示,只有12个NBS-LRR类基因在PGSC的转录组数据中完全没有检测到表达,不同NBS-LRR类基因转录水平不同,一部分NBS-LRR类基因有较高的生物胁迫和激素刺激应激性。分析了这433个基因的编码区域的单核苷酸多态性(SNP)位点,SNP标记作为在高通量测序和生物大数据分析中最常使用的遗传标记,有助于鉴定NBS-LRR基因在不同马铃薯品种中的实际抗病性。
关键词 马铃薯;晚疫病;抗性基因
中图分类号 Q37 文献标识码 A 文章编号 0517-6611(2023)10-0079-06
doi:10.3969/j.issn.0517-6611.2023.10.017
Abstract In this paper, 433 NBSLRR genes were screened from the Solanum tuberosum reference genome, and the distribution of these genes in the genome were mapped, showing that some of these genes were distributed in clusters. Only 12 NBSLRR genes were not detected in all of the transcriptome analysis data of PGSC. The expression levels of different NBSLRR genes were different, and some NBSLRR genes had higher biological and hormonal stimulation irritability. The single nucleotide polymorphism (SNP) sites in the coding regions of these 433 genes were also analyzed. As the most commonly used genetic markers in highthroughput sequencing and biological big data analysis, SNP markers are helpful for the identification of actual disease resistance of NBSLRR genes in Solanum tuberosum varieties.
Key words Solanum tuberosum;Phytophthora infestans (Mont.) DE Bary;Resistance (R) gene
马铃薯(Solanum tuberosum)适应性广,丰产性好,营养丰富,经济效益高,已成为世界上继水稻、小麦的第三大粮食作物[1]。但在生产中经常受到其第一大病害——晚疫病的危害造成产量严重降低。晚疫病是由致病疫霉菌[Phytophthora infestans(Mont.)de Bary.]引起,特别是由于病原菌A2交配型的出现,与原A1交配型有性杂交,发生变异,导致最初的抗病马铃薯品种失去抗性[2-3]。
目前已经发现多个马铃薯晩疫病抗病基因(R基因),包括克隆自Solanum demissum中,位于第5号染色体上的R1[4],第4号染色体上的R2[5],第11号染色体上的R3a[6]和R3b[7],第9号染色体上的R8[8];克隆自Solanum bulbocastanum,位于第8号染色体上的Rpi-blb1(RB)[9-10],第6号染色体上的Rpi-blb2[11],第9号染色体上的Rpi-blb3和第4号染色体上的Rpi-abpt[5];来源于Solanum venturii位于第9号染色体上Rpi-vnt1.1、Rpi-vnt1.2和Rpi-vnt1.3[12];来源于Solanum stoloniferum位于第8号染色体上Rpi-sto1和Rpi-pta1[13-14];来源于Solanum americanum位于第4号染色体上Rpi-amr3i[15]。已鑒定出的R基因都拥有NBS-LRR(Nucleotidebinding site and leucinerich repeat)结构,因此可以利用基因结构上的保守型从参考基因组中筛选潜在的抗性基因[16-17]。目前已在多个植物的参考基因组中筛选出NBS-LRR类基因,包括重要的作物水稻[18]、小麦[19]、玉米[20]、大豆[21]、棉花[22]、十字花科[23]、黄瓜[24]、模式生物-拟南芥[25]以及茄科植物辣椒、番茄[26]和茄子[27]等。虽然马铃薯晩疫病抗病基因能提供抗病性,但单个R基因导入带来的抗性易被变异的致病疫霉菌克服,如将RD基因导入而育成的栽培种Biogold,在田间种植不到1年即被晚疫病菌克服[28],但通常认为引入多个R基因能够增强晚疫病抗病表现[29]。因此,尽可能多地筛选出晩疫病抗病基因,有助于马铃薯晚疫病抗性育种。
高通量测序技术提高了人们对作物遗传学的理解。由于普通栽培马铃薯基因组是高度杂合的同源4倍体的特性。马铃薯基因组测序协会(potato genome sequencing consortium,PGSC)首先通过对一个纯合的双单倍体(DM1-3 516 R44,DM)马铃薯品种测序得到了马铃薯参考基因组,参考基因组提供了详细的马铃薯基因座信息,因此可以通过序列比对在参考基因组中查找具有保守序列和结构域的基因[30]。并且利用参考基因组对一个(杂合二倍体RH89-039-16,RH)马铃薯品种重测序,基因分型得到了两者之间的单核苷酸多态性位点(SNP)。PGSC还利用参考基因组对DM的16种组织包括:心皮、花瓣、萼片、雄蕊、花、果肉、未成熟果实、成熟果实、叶柄、叶、匍匐茎、未成熟块茎、成熟块茎、愈伤组织、根和芽的转录组,以及11种经处理包括:盐、甘露醇和热胁迫,4种激素[IAA(吲哚乙酸)、BAP(6-苄基腺嘌呤)、GA3(赤霉酸)和ABA(脱落酸)]处理,4种生物胁迫[接种晚疫病菌、2种诱导植物抗病的因子苯并噻二唑(BTH)和DL-氨基丁酸(BABA)处理以及受伤叶片]后转录组表达水平的变化情况处理进行了转录组测序。并且作为对DM转录组数据的补充和对照,PGSC还对杂合二倍体品种RH的花、雄蕊、根、叶、水分胁迫叶、叶柄、茎尖、茎、匍匐茎、未成熟块茎、成熟块茎、块茎髓、块茎皮、块茎芽、块茎皮质和整株16种组织的转录组进行了测序。转录组测序数据经随后的RNA定量验证显示出很高的准确性[30-31]。因此,可以利用R基因的NBS-LRR结构特性,分析马铃薯参考基因组种的NBS-LRR类基因,并通过转录组数据分析这些基因在不同组织和处理条件下的表达特性,以及利用基因组序列信息,分析分子标记信息,为鉴定各基因的实际抗病性鉴定打下理论基础。
1 材料与方法
1.1 马铃薯NBS-LRR类基因的鉴定
使用BLASTp工具(http://spuddb.uga.edu/blast.shtml),以期望值(E-value=1)为阈值,用已知的R基因的蛋白序列作为查询序列,以及隐马模型(Hidden Markov Model,HMM)[32],以Pfam:PF00931作为查询序号,以默认参数从马铃薯参考基因组(V 4.03)中鉴定(http://spuddb.uga.edu/integrated_searches.shtml)。利用SMART检索基因的蛋白结构域(http://smart.emblheidelberg.de/)[33];以0.9为阈值,利用 COILS 程序检索基因的CC结构域(http://www.ch.embnet.org/software/COILS_form.html)[34]。基因的氨基酸序列利用Clustal Omega在线服务器进行多序列比对,利用Neighbor-Joining参数进过计算1 000次生成树图(http://www.ebi.ac.uk/Tools/msa/clustalo/)。
1.2 马铃薯NBS-LRR类基因的表达分析
利用马铃薯基因组测序协会(potato genome sequencing consortium,PGSC)的Illumina RNA 测序数据分析NBS-LRR类基因在不同组织和处理方式下的表达模式[30-31]。转录组数据的表达丰度用每百万个映射上的片段数中映射到外显子中的每1 000个碱基上的片段个数(fragments per kilobase of exon model per million mapped reads,FPKM)值表示。对不同组织内NBS-LRR类基因表达水平的热图(Heat map)进行构建时,FPKM 值首先经过以2为底数的对数进行转化。构建生物胁迫以及激素处理时NBS-LRR类基因表达水平变化的热图时,首先计算与对照的对比值,以log2比值的相对值≥1确定为差异表达基因[31]。热图使用 MeV 4.9.0 软件进行构建[35]。相关性分析用IBM SPSS Statistics 22 软件进行(SPSS GmbH Software,Munich,Germany)。
1.3 马铃薯NBS-LRR类基因编码序列的SNP标记分析
NBS-LRR类基因编内的SNP标记信息来源于以DM为参考基因组,RH重测序信息,Sol-CAP对于6个常见栽培种的转录组测序信息,以及对大西洋(Atlantic)和Superior的重测序信息[6,36]。
2 结果与分析
2.1 马铃薯NBS-LRR类基因的鉴定
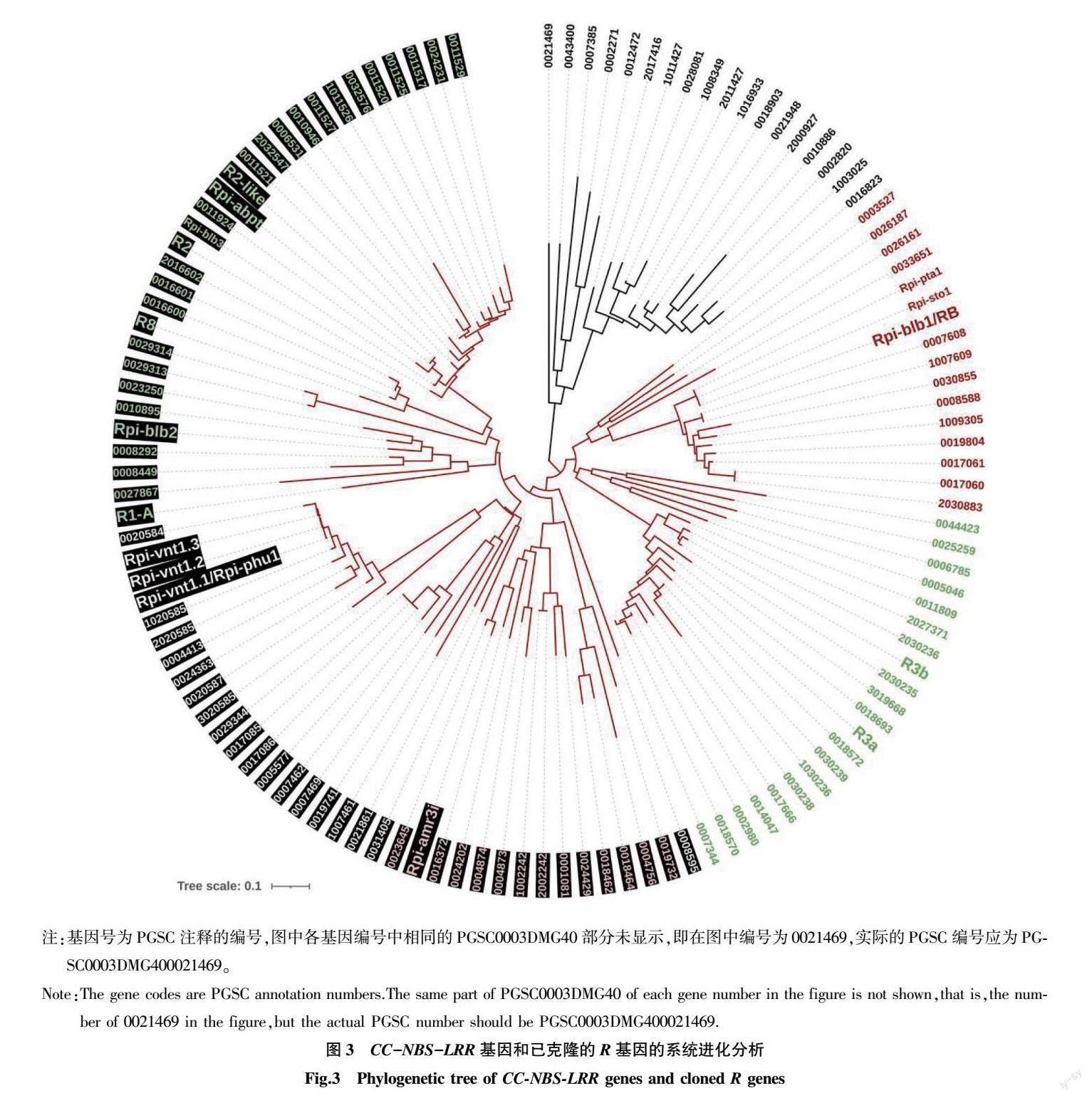
从马铃薯参考基因组中鉴定得到433个NBS-LRR类基因,其中366个基因能够定位到12个染色体中。在这些基因中的142个基因间的距离小于1.0 mb,其中44个基因间的距离小于10 kb。21个基因定位于第4染色体上2.0 mb的染色体区域,12个基因定位于第5染色体上1.5 mb的染色体区域,22个基因定位于第11染色体上2.0 mb的染色体区域,说明这些基因的其中一部分成簇分布(图1)。433个NBS-LRR类基因中的103个基因蛋白编码序列的N端含有一个CC(coiled-coil)结构域,这与已发现的晚疫病抗性基因的CC-NBS-LRR蛋白结构域相同(图2)。进一步利用已知的R基因和CC-NBS-LRR基因的氨基酸序列进行比对并绘制系统进化树。进化树分成2个部分(黑色和红色),其中黑色部分没有已克隆的R基因。红色部分的进化树又可分成2个部分(基因编号突出显示和未突出显示的部分),这2部分内部又可分成2~3个小组(基因编号不同颜色字体显示)。Rpi-blb1(RB)与15个CC-NBS-LRR基因聚集成一个小组,R3a和R3b 与19个CC-NBS-LRR基因亲缘关系较近,Rpi-amr3i与14个CC-NBS-LRR基因聚集成一个小组,Rpi-vnt1.1、Rpi-vnt1.2和Rpi-vnt1.3与17个CC-NBS-LRR基因聚集成一个小组,其他7个已克隆的R基因与23个CC-NBS-LRR基因亲缘关系较近(图3)。
2.2 马铃薯NBS-LRR类基因的表达分析
利用PGSC在马铃薯不同器官和发育阶段以及在胁迫处理和激素刺激时的不同转录组测序所产生的数据,包括:DM的16种组织内基因的表达水平,DM的11种包括盐胁迫、激素刺激、生物胁迫处理后转录组表达水平的变化情况,以及以DM为参考基因组,RH的16种组织内基因的表达模式。分析NBS-LRR类基因的表达。转录组数据分析显示,只有12个NBS-LRR类基因在所有的组织或处理中完全没有检测到表达(图2)。
PGSC分别在DM和RH的16种不同组织中进行了转录组分析,提取了其中易受晚疫病侵染影响的组织的转录组表达数据,进一步分析了433个NBS-LRR类基因在不同组织和样品间的表达模式。在DM的6种组织中(根、叶、叶柄、匍匐茎、不成熟块茎和成熟块茎),433个NBS-LRR类基因,表达量FPKM均值最大的10个基因分别是PGSC0003DMG400007999、PGSC0003DMG400009635、PGSC0003DMG400007469、PGSC0003DMG400003527、PGSC0003DMG402018257、PGSC0003DMG400018464、PGSC0003DMG400031405、PGSC0003DMG400020587、PGSC0003DMG400033159和PGSC0003DMG400007471,其表达量的FPKM值在52.67~11.60。有57个基因在DM的6种组织中没有检测到表达,208个基因在这6种组织中至少有1种组织中没有检测到表达。376个基因在至少DM的6种组织之一中检测到表达,反映表达量在6种组织间离散程度的标准差(σ),最大为34.13,平均为1.76。在RH的8种组织中(根、叶、叶柄、植株、茎、匍匐茎、不成熟块茎和成熟块茎),433个之间NBS-LRR类基因,表达量均值最大的10个基因分别是PGSC0003DMG400007999、PGSC0003DMG400007469、PGSC0003DMG400031405、PGSC0003DMG400013490、PGSC0003DMG400029505、PGSC0003DMG400005335、PGSC0003DMG400007462、PGSC0003DMG400018428、PGSC0003DMG400018429和PGSC0003DMG400026187,其表达量的FPKM值在60.27~12.58。有32个基因在RH的8种组织中没有检测到表达,179个基因在这8种组织中至少有1种组织中没有检测到表达。401个基因在至少RH的8种组织之一检测到表达,反映表达量FPKM值在8种组织间离散程度的标准差(σ),最大为62.45,平均为1.57。结果显示,相同的NBS-LRR类基因在DM或RH的不同组织间的表达水平有一定的差异,但整体上差异并不明显,即组织特异性不明显。在DM和RH组织之间,相同基因表达量的FPKM值均值,其中在DM中大于RH中的基因有126个,在RH中大于DM中的基因有283个,说明相同的NBS-LRR类基因在DM和RH之间,整体上基因在RH中表达水平略高于在DM中。433个NBS-LRR基因在DM和RH组织间表达水平的Pearson相關系数r=0.600(P<0.01),有显著的正相关关系,说明NBS-LRR基因在DM和RH的组织中有类似的表达模式。
生物胁迫处理包括接种马铃薯晚疫病菌和2种诱导植物抗病的因子[苯并噻二唑(BTH,100 mg/mL)以及DL-氨基丁酸(BABA,2 mg/mL)]处理24 h/48 h/72 h后混合样本的表达丰度。参加分析的数据为计算每个处理相对于对照的相对表达丰度。从结果中可知,在接种晚疫病菌处理后,有32个基因表达量显著上升(基因表达量log2比值≥1,即处理后的基因表达量是对照的基因表达量的2倍及以上);4个基因(PGSC0003DMG400011521、PGSC0003DMG400011906、PGSC0003DMG400033161和PGSC0003DMG400009307)的基因表达量log2比值>2。在接受诱导植物抗病的BTH因子处理后,有36个基因表达量显著上升,3个基因(PGSC0003DMG400011906、PGSC0003DMG400010527和PGSC0003DMG400044423)的表达量log2比值>2。在BABA因子处理后,有179个基因表达量显著上升,32个基因的表达量log2比值>2,3个基因(PGSC0003DMG401007609、PGSC0003DMG403015877和PGSC0003DMG400028081)的表达量log2比值>4(图2)。在3种生物胁迫方式处理后,有4个基因(PGSC0003DMG400011521、PGSC0003DMG400011906、PGSC0003DMG400033161和PGSC0003DMG400044423)至少在接受2种处理后基因表达量log2比值>2。
有研究显示,晚疫病抗性基因的表达受到激素的影响[37]。为研究激素对NBS-LRR类基因的表达的影响,分析整株植物经IAA(吲哚乙酸)10 mmol /L、BAP(6-苄基腺嘌呤)10 mmol /L、GA3(赤霉酸)50 mmol /L、ABA(脱落酸)50 mmol /L 处理24 h后的相对表达丰度。在IAA刺激后,有80个基因表达量显著上升(基因表达量log2比值≥1,即处理后的基因表达量是对照的基因表达量的2倍及以上),4个基因(PGSC0003DMG400020741、PGSC0003DMG400011521、PGSC0003DMG400029220和PGSC0003DMG400001989)的基因表达量log2比值>2。在BAP处理后,有66个基因表达量显著上升,15个基因表达量log2比值>2,2个基因(PGSC0003DMG400010612和PGSC0003DMG400026433)的表达量log2比值>4。在GA3刺激后,有159个基因表达量显著上升,17个基因表达量log2比值>2,2个基因(PGSC0003DMG 400020741和PGSC0003DMG400019804)的表达量log2比值>4。在ABA处理后,有51个基因表达量显著上升,2个基因(PGSC0003DMG400020741和PGSC0003DMG400011521)的表达量log2比值>2。在4种激素刺激后,有8个基因(PGSC0003DMG400001989、PGSC0003DMG400002427、PGSC0003DMG400003527、PGSC0003DMG400011521、PGSC0003DMG400019804、PGSC0003DMG400020587、PGSC0003DMG400020741和PGSC0003DMG400026433)至少在接受2种处理后基因表达量log2比值>2(图2)。
2.3 马铃薯NBS-LRR类基因编码序列的SNP标记分析
SNP标记是DNA中的单碱基变异造成的,在该研究中,以双单倍体DM为参考基因组,利用杂合二倍体RH重测序信息,Sol-CAP对于6个常见栽培种的转录组测序信息,以及对栽培种大西洋(Atlantic)和Superior的重测序信息,分析在433个NBS-LRR类基因编码序列内的SNP标记。结果显示,在以上9个品种的序列信息内,只有19个NBS-LRR类基因的编码区域内没有SNP,134个NBS-LRR类基因的编码区域内有1~10个SNP,67个NBS-LRR类基因的编码区域内有超过50个SNP(图2)。同时,还分析了NBS-LRR类基因编码序列内SNP的数量是否影响其表达,结果显示没有相关性。
3 讨论与结论
目前已发现的晚疫病抗病(R)基因都是NBS-LRR家族基因,利用已测序完成的参考基因组序列信息,可以初步筛选潜在的抗性基因。目前已在多个植物基因组中鉴定出NBS-LRR家族候选基因,包括重要的作物水稻[12]、小麦[13]、玉米[14]、大豆[15]、棉花[16]、十字花科[17]、黄瓜[18]以及模式生物-拟南芥[19]。在茄科植物中,从辣椒的参考基因组中鉴定到了305个 NBS-LRR类基因,从番茄的参考基因组中鉴定到了255个NBS-LRR类基因[20],从茄子的参考基因组中鉴定到了245个NBS-LRR类基因[21]。该研究从马铃薯参考基因组中鉴定出433个NBS-LRR类基因(图1)。相较于其他茄科植物,从马铃薯参考基因组中鉴定出更多的NBS-LRR类基因,可能是由于更多的基因组倍性变化,如普通栽培种马铃薯的基因组是四倍体,而野生马铃薯的基因组是二倍体至六倍体,导致的基因组更多变异的累加,并分化出不同的NBS-LRR类基因。这些NBS-LRR类基因的其中一部分成簇分布(图1),成簇分布的基因,由于连锁效应,将更易于在杂交育种的过程中,一次性转移更多的NBS-LRR类基因进行品种改良,而选育晚疫病抗性马铃薯品种需要更多的R基因累加。
基因发挥功能首先必须转录成mRNA,在该研究中利用PGSC的转录组数据,包括DM的16种组织内基因的表达水平,DM对11种包括盐胁迫、激素刺激、生物胁迫处理后转录组表达水平的变化情况,以及以DM为参考基因组,RH的16种组织内基因的表达模式[30-31]。转录组数据分析显示,只有12个NBS-LRR类基因在所有的组织或处理中完全没有检测到表达。进一步分析转录组数据,在易受晚疫病侵染影响的DM的6种组织和RH的8种组织中NBS-LRR基因的表达情况中,不同的NBS-LRR基因表达水平不同,但相同的NBS-LRR基因在DM或RH的不同组织间的表达水平有一定差异,但整体上差异并不明显,即组织特异性不明显。相同的NBS-LRR基因整体上在RH中表达水平略高于在DM中的表达水平。433个NBS-LRR基因在DM和RH组织间表达水平有显著的正相关关系,说明NBS-LRR基因在DM和RH中表達模式类似。同时,部分NBS-LRR基因在生物胁迫处理和激素刺激后,表达水平显著上升,有明显的应激性。
SNP標记作为在高通量测序和生物大数据分析中最常使用的遗传标记,在该研究中分析了433个NBS-LRR基因的编码区域内的SNP位点(图2),已知的SNP标记,可以利用多态性位点及侧翼序列,通过如KASP(Kompetitive Allele Specific PCR)高通量基因分型技术,实现在不同品种中对该位点较快速的基因分型[38],有助于鉴定NBS-LRR基因在不同马铃薯品种中的实际抗病性。根据选择性清除理论,在受选择的基因组区域,其多态性会降低,表现为SNP标记数目减少[39],尽管研究结果显示NBS-LRR基因编码区域内的SNP数量的多少与其表达不相关,但67个NBS-LRR基因的编码区域内存在50个以上的SNP,可能代表基因组的这部分区域重组频繁,对基因的功能也可能产生一定的影响。
参考文献
[1] BHASKAR P B,VENKATESHWARAN M,WU L,et al.Agrobacteriummediated transient gene expression and silencing:A rapid tool for functional gene assay in potato[J].PLoS One,2009,4(6):1-8.
[2] FRY W E,GOODWIN S B.Resurgence of the Irish potato famine fungus[J].Bioscience,1997,47:363-371.
[3] FRY W.Phytophthora infestans:The plant(and R gene)destroyer[J].Molecular plant pathology,2008,9(3):385-402.
[4] BALLVORA A,ERCOLANO M R,WEISS J,et al.The R1 gene for potato resistance to late blight(Phytophthora infestans)belongs to the leucine zipper /NBS /LRR class of plant resistance genes[J].Plant journal,2002,30(3):361-371.
[5] LOKOSSOU A A,PARK T H,VAN ARKEL G,et al.Exploiting knowledge of R/Avr genes to rapidly clone a new LZNBSLRR family of late blight resistance genes from potato linkage group IV[J].Molecular plantmicrobe interactions,2009,22(6):630-641.
[6] HUANG S W,VAN DER VOSSEN E A,KUANG H H,et al.Comparative genomics enabled the isolation of the R3a late blight resistance gene in potato[J].Plant journal,2005,42(2):251-261.
[7] LI G C,HUANG S W,GUO X,et al.Cloning and characterization of R3b;members of the R3 superfamily of late blight resistance genes show sequence and functional divergence[J].Molecular plantmicrobe interactions,2011,24(10):1132-1142.
[8] VOSSEN J H,VAN ARKEL G,BERGERVOET M,et al.The Solanum demissum R8 late blight resistance gene is an Sw5 homologue that has been deployed worldwide in late blight resistant varieties[J].Theoretical and applied genetics,2016,129(9):1785-1796.
[9] VAN DER VOSSEN E,SIKKEMA A,HEKKERT B T,et al.An ancient R gene from the wild potato species Solanum bulbocastanum confers broadspectrum resistance to Phytophthora infestans in cultivated potato and tomato[J].Plant journal,2003,36(6):867-882.
[10] SONG J Q,BRADEEN J M,NAESS S K,et al.Gene RB cloned from Solanum bulbocastanum confers broad spectrum resistance to potato late blight[J].Proc Natl Acad Sci USA,2003,100(16):9128-9133.
[11] VAN DER VOSSEN E A,GROS J,SIKKEMA A,et al.The Rpiblb2 gene from Solanum bulbocastanum is an Mi1 gene homolog conferring broadspectrum late blight resistance in potato[J].Plant journal,2005,44(2):208-222.
[12] FOSTER S J,PARK T H,PEL M,et al.Rpivnt1.1,a Tm22 homolog from Solanum venturii,confers resistance to potato late blight[J].Molecular plantmicrobe interactions,2009,22(5):589-600.
[13] VLEESHOUWERS V G A A,RIETMAN H,KRENEK P,et al.Effector genomics accelerates discovery and functional profiling of potato disease resistance and Phytophthora infestans avirulence genes[J].PloS One,2008,3(8):1-10.
[14] WANG M Q,ALLEFS S,VAN DEN BERG R G,et al.Allele mining in Solanum:Conserved homologues of Rpiblb1 are identified in Solanum stoloniferum[J].Theoretical and applied genetics,2008,116(7):933-943.
[15] WITEK K,JUPE F,WITEK A I,et al.Accelerated cloning of a potato late blightresistance gene using RenSeq and SMRT sequencing[J].Nature biotechnol,2016,34(6):656-660.
[16] MARTIN G B,BOGDANOVE A J,SESSA G.Understanding the functions of plant disease resistance proteins[J].Annual review of plant biology,2003,54:23-61.
[17] VAN DER BIEZEN E A,JONES J D.The NBARC domain:A novel signalling motif shared by plant resistance gene products and regulators of cell death in animals[J].Current biology,1998,8(7):R226-R227.
[18] LIU W,GHOURI F,YU H,et al.Genome wide resequencing of newly developed Rice Lines from common wild rice(Oryza rufipogon Griff.)for the identification of NBSLRR genes[J].PLoS One,2017,12(7):1-19.
[19] GU L J,SI W N,ZHAO L N,et al.Dynamic evolution of NBSLRR genes in bread wheat and its progenitors[J].Molecular genetics genomics,2015,290(2):727-738.
[20] CHENG Y,LI X Y,JIANG H Y,et al.Systematic analysis and comparison of nucleotidebinding site disease resistance genes in maize[J].FEBS journal,2012,279(13):2431-2443.
[21] KANG Y J,KIM K H,SHIM S,et al.Genomewide mapping of NBSLRR genes and their association with disease resistance in soybean[J].BMC plant biology,2012,12:1-13.
[22] KHAN A M,KHAN A A,AZHAR M T,et al.Comparative analysis of resistance gene analogues encoding NBSLRR domains in cotton[J].Journal of the science of food and agriculture,2016,96(2):530-538.
[23] ZHANG Y M,SHAO Z Q,WANG Q,et al.Uncovering the dynamic evolution of nucleotidebinding siteleucinerich repeat(NBSLRR)genes in Brassicaceae[J].Journal of integrative plant biology,2016,58(2):165-177.
[24] WAN H J,YUAN W,BO K J,et al.Genomewide analysis of NBSencoding disease resistance genes in Cucumis sativus and phylogenetic study of NBSencoding genes in Cucurbitaceae crops[J].BMC genomics,2013,14:1-15.
[25] MONDRAGNPALOMINO M,MEYERS B C,MICHELMORE R W,et al.Patterns of positive selection in the complete NBSLRR gene family of Arabidopsis thaliana[J].Genome research,2002,12(9):1305-1315.
[26] SHAO Z Q,XUE J Y,WU P,et al.Largescale analyses of angiosperm nucleotidebinding siteleucinerich repeat genes reveal three anciently diverged classes with distinct evolutionary patterns[J].Plant physiology,2016,170(4):2095-2109.
[27] WEI H W,LIU J,GUO Q W,et al.Genomic organization and comparative phylogenic analysis of NBSLRR resistance gene family in Solanum pimpinellifolium and Arabidopsis thaliana[J/OL].Evolutionary bioinformatics,2020,16[2022-03-17].https://doi.org/10.1176934320911055.
[28] HERMSEN J G TH,RAMANNA M S.Doublebridge hybrids of Solanum bulbocastanum and cultivars of Solanum tuberosum[J].Euphytica,1973,22(3):457-466.
[29] HAVERKORT A J,BOONEKAMP P M,HUTTEN R,et al.Durable late blight resistance in potato through dynamic varieties obtained by cisgenesis:Scientific and societal advances in the DuRPh project[J].Potato research,2016,59(1):35-66.
[30]The Potato Genome Sequencing Consortium.Genome sequence and analysis of the tuber crop potato[J].Nature,2011,475(7355):189-195.
[31] MASSA A N,CHILDS K L,LIN H N,et al.The transcriptome of the reference potato genome Solanum tuberosum Group Phureja clone DM13516R44[J].PLoS One,2011,6(10):1-8.
[32] ELGEBALI S,MISTRY J,BATEMAN A,et al.The Pfam protein families database in 2019[J].Nucleic acids research,2019,47:D427-D432.
[33] LETUNIC I,KHEDKAR S,BORK P.SMART:Recent updates,new developments and status in 2020[J].Nucleic acids research,2021,49(D1):D458-D460.
[34] LUPAS A,VAN DYKE M,STOCK J.Predicting coiled coils from protein sequences[J].Science,1991,252(5009):1162-1164.
[35] SAEED A I,SHAROV V,WHITE J,et al.TM4:A free,opensource system for microarray data management and analysis[J].Biotechniques,2003,34(2):374-378.
[36] HAMILTON J P,HANSEY C N,WHITTY B R,et al.Single nucleotide polymorphism discovery in elite North American potato germplasm[J].BMC genomics,2011,12:1-12.
[37] TURNBULL D,YANG L N,NAQVI S,et al.RXLR effector AVR2 upregulates a brassinosteroidresponsive bHLH transcription factor to suppress immunity[J].Plant physiology,2017,174(1):356-369.
[38] ALVAREZFERNANDEZ A,BERNAL M J,FRADEJAS I,et al.KASP:A genotyping method to rapid identification of resistance in Plasmodium falciparum[J].Malar journal,2021,20(1):1-8.
[39] NIELSEN R,WILLIAMSON S,KIM Y,et al.Genomic scans for selective sweeps using SNP data[J].Genome research,2005,15(11):1566-1575.
