山楂属植物叶绿体基因组特征与密码子偏好性分析
2023-06-08赵振宁孙浩田宋雨茹余潇
赵振宁 孙浩田 宋雨茹 余潇

摘要: 為明确山楂属植物叶绿体基因组结构与编码蛋白质的基因密码子偏好性特征,本研究利用第二代高通量测序技术对云南山楂[Crataegus scabrifolia (Franch.) Rehd.]的叶绿体基因组进行测序、组装和注释,并对山楂属11个种植物的叶绿体基因组结构、遗传多样性以及密码子偏好性进行了分析。结果显示,山楂属植物的叶绿体基因组长度为159 607~159 875 bp,G+C含量为36.6%~36.7%,为标准的四分体结构,G+C含量和结构变异均保守,边界扩张收缩稳定,未发现基因组的倒置和重排现象,11个种植物的简单重复序列和离散重复序列的种类和数量存在一定的差异。综合中性绘图分析、有效密码子数分析(ENC-plot)、奇偶校验分析(PR2-plot)和对应性(COA)分析的结果,发现山楂属植物叶绿体基因组密码子使用不但受到碱基突变的影响,还受到选择压力的深刻影响。对叶绿体基因组的最优密码子进行筛选,最优密码子数量为17~20个,其中C. kansuensis、C. oresbia、C. pinnatifida的最优密码子数量最多,C. marshallii的最优密码子数量最少,分析它们的最优密码子数据发现,山楂属植物的最优密码子大多以A或U作为第三位碱基。基于CDS(蛋白质编码序列)和叶绿体全基因组构建的系统发育关系既具有一定的相似性,也存在一些差异。本研究结果为山楂属植物的系统发育研究和分子标记开发等工作提供了参考依据。
关键词: 山楂属;叶绿体基因组;密码子偏好性;系统进化
中图分类号: S661.5 文献标识码: A 文章编号: 1000-4440(2023)02-0504-14
Chloroplast genome characteristics and codon usage bias analysis of Crataegus L.
ZHAO Zhen-ning1, SUN Hao-tian2, SONG Yu-ru1, YU Xiao3
(1.College of Forestry, Southwest Forestry University, Kunming 650224, China;2.College of Ecology and Environment, Southwest Forestry University, Kunming 650224, China;3.School of Architecture, Hubei Engineering University, Xiaogan 432000, China)
Abstract: In order to clarify the chloroplast genome structure and codon usage bias of Crataegus, this study used the next-generation sequencing to sequence, assemble and annotate the chloroplast genome of Crataegus scabrifolia (Franch.) Rehd., and analyzed the chloroplast genome structure, genetic diversity and codon preference of 11 species of Crataegus. The results showed that the length of chloroplast genome was between 159 607 bp and 159 875 bp, the G+C content and structural variation were conservative, the G+C content was between 36.6% and 36.7%, the boundary expansion and contraction were stable, no inversion and rearrangement of the genome were found, and there were differences in the type and number of simple sequence repeats and interspersed repeated sequences. Based on the results of neutrality plot analysis, ENC-plot, PR2-plot and correspondence analysis, it was found that the chloroplast genome codon usage in Crataegus was not only affected by base mutation, but also by selective pressure. The optimal codons of the chloroplast genome were screened, and the optimal number of codons was between 17 and 20. C. kansuensis, C. oresbia, and C. pinnatifida had the largest number of optimal codons, and C. marshallii had the least number of optimal codons. The analysis of their optimal codon data revealed that the optimal codons of Crataegus mostly used A or U as the third base. The phylogenetic relationships constructed based on protein coding sequence and complete chloroplast genome had certain similarities and differences. The results of this study can provide a reference for the phylogenetic research and molecular marker development of Crataegus.
Key words: Crataegus L.;chloroplast genome;codon usage bias;system evolution
山楂属(Crataegus L.)为蔷薇科中起源相对古老的属,多为小乔木或落叶灌木,主要分布于温带地区。山楂属植物有着非常高的经济价值,研究结果表明,山楂作为果树在中国的种植历史可追溯至汉代[1]。山楂的果实含有丰富的营养物质,具有健胃消食、抗菌消炎等功效,是一种优良的水果[2]。除了作为经济果树,山楂还是一类出色的园林景观植物和街道绿化树种。通常认为,山楂属中有18个种原产于中国,山楂属植物中广泛存在的无融合生殖和种间杂交现象使其外形特征发生了高度变异[3],进而为山楂属植物的传统分类学鉴定造成困难。
叶绿体是植物细胞中重要的细胞器之一,对于研究植物体的光合作用和生长发育具有非常重要的意义。叶绿体基因组是独立于核基因组的母系遗传,其核苷酸置换率与核基因组及线粒体基因组相比更适宜应用于多层次的系统发育研究[4]。随着第二代高通量测序技术的不断完善,针对叶绿体基因组的报道也逐渐增多,目前的研究结果表明,陆地高等植物的叶绿体基因组长度一般介于120~200 kb,包含大单拷贝区(LSC)、小单拷贝区(SSC)、反向重复区a(IRa)和反向重复区b(IRb)。密码子偏好性是指编码相同氨基酸的同义密码子频率存在差异[5],这种现象普遍出现在所有原核生物和真核生物中[6]。一般来说,密码子使用模式能够反映基因组的起源和进化模式,不同的基因组有其独特的密码子使用偏好性,这也使得解释这种偏好性目前还存在一定的困难[7-8]。
山楂属植物具有出色的经济价值和科研价值,目前已有许多针对山楂属植物的相关研究。例如,有许多学者围绕山楂属植物的营养价值进行了相关研究,均发现其有着良好的营养价值和抗氧化活性[9-12],在分子层面,张枭等[13]利用SSR分子标记构建了部分山楂属植物的分子条形码,为山楂属植物的资源鉴定提供了分子层面的手段,Liston等[3]基于叶绿体基因组和257个核基因组对山楂属植物亚属间的杂交状况进行了评估,证实了杂交在山楂进化中的重要作用。具体到叶绿体基因组层面,近年来,针对山楂属植物叶绿体基因组的研究正逐渐被重视,部分山楂属植物的叶绿体基因组数据相继被发表在国家生物技术信息中心(National Center for Biotechnology Information, NCBI)公共数据库中,也有学者对其叶绿体基因组进行了属内的比较分析[14-15]。然而,目前针对山楂属植物叶绿体基因组特征和密码子偏好性的综合分析相对较少。本研究拟通过对云南山楂叶绿体基因组的测序、组装和注释,综合分析山楂属11个种的植物叶绿体基因组特征、密码子偏好性、最优密码子和系统发育关系,深入研究山楂属植物的叶绿体基因组特征,弥补目前对于山楂属植物密码子特征和偏好性研究的空白。本研究旨在为山楂属植物的叶绿体基因组特征、系统发育关系和密码子偏好性研究提供新的参考依据,以期为山楂属植物的育种和分子标记研究提供参考。
1 材料与方法
1.1 试验材料
本研究所使用的新鲜植物叶片采集于云南省大理白族自治州洱源县罗平山(99°52′19.15″E, 25°59′53.34″N,海拔2 105 m),經西南林业大学标本馆树木学教研室李双智副教授鉴定为蔷薇科山楂属植物云南山楂[Crataegus scabrifolia (Franch.) Rehd.]。使用改良过的CTAB(十六烷基三甲基溴化铵)法[16]从使用硅胶干燥的叶片中提取DNA,提取后的DNA送至天津诺禾致源生物科技有限公司进行叶绿体基因组测序,使用GetOrganelle软件 [17]组装得到完整的叶绿体基因组,并使用拼接路径可视化软件Bandage[18]验证其成环性。以山楂[Crataegus pinnatifida(NC_065486)]叶绿体基因组为参考,使用CPGAVAS2在线工具(http://www.herbalgenomics.org/cpgavas/)[19]对云南山楂叶绿体基因组进行注释,并使用Geneious Prime软件[20]对其进行手动调整。注释过的云南山楂叶绿体基因组上传到GenBank公共数据库,登录号为OP021659,其余10个山楂属植物叶绿体基因组下载于NCBI公共数据库(https://www.ncbi.nlm.nih.gov/)(表1)。
1.2 试验方法
1.2.1 重复序列分析 简单重复序列(Simple sequence repeat,SSR)在植物叶绿体基因组中有着广泛分布,其作为一种重要的分子标记常被用作鉴定植物品种和构建DNA 指纹图谱[21]。使用MISA-web(http://webblast.ipk-gatersleben.de/misa/)对山楂属植物简单重复序列的种类和数量进行在线分析[22],将单核苷酸、二核苷酸、三核苷酸、四核苷酸、五核苷酸、六核苷酸参数分别设置为10、5、4、3、3、3,相邻SSR间的最小距离为100 bp。
使用REPuter在线工具(https://bibiserv.cebitec.uni-bielefeld.de/reputer)分别鉴定11种山楂属植物的离散重复序列[23],设置参数:海明距离(Hamming distance)为3,鉴定类型选择正向重复序列(Forward repeat, F)、回文重复序列(Palindromic repeat, P)、反向重复序列(Reverse repeat, R)和互补重复序列(Complement repeat, C)4种,最小重复长度30 bp,最大重复长度300 bp。
1.2.2 边界扩张收缩分析 叶绿体基因组为环状结构,分为4个区域,分别为大单拷贝区(LSC)、小单拷贝区(SSC)、反向重复区a(IRa)和反向重复区b(IRb),其中反向重复区相对比较保守,其收缩与扩张会影响叶绿体基因组 G+C 含量和基因组大小,边界扩展和收缩能够展现植物的遗传进化[24];分析叶绿体基因组区域边界的信息,对揭示叶绿体基因组的结构差异和进化关系具有重要的参考价值[25]。使用在线工具CPJSdraw(http://cloud.genepioneer.com:9929)对注释过的山楂属植物叶绿体基因组边界可视化,分析其边界的扩张收缩情况。
1.2.3 共线性比较分析 以山楂属11个种的植物叶绿体基因组为研究对象,利用 MAUVE(http://darlinglab.org/mauve/mauve.html)工具对多重基因组的保守区域、局部共线性和基因组重排倒置现象进行鉴定,用以阐述山楂属植物的叶绿体在物种演化过程中发生的结构变异事件[26]。
1.3 密码子偏好性分析
1.3.1 密码子相关参数的计算 使用Geneious Prime软件手动提取每个山楂属植物叶绿体基因组中的蛋白质编码序列(Coding sequence, CDS),由于編码长度较短的蛋白质的基因会使密码子偏好性的数据存在较大的估计误差,因此在统计密码子偏好性时,常去除长度在300 bp以下的序列,从而避免产生统计误差[27],本研究筛选了山楂属植物叶绿体基因组中具有代表性的48个CDS。利用CUSP在线工具(http://www.Bioin formatics.nl/emboss-explorer/)和Condon W 1.4.2统计得到了叶绿体基因组的相对同义密码子使用度(RSCU),密码子第一、第二和第三位的G+C含量(GC1、GC2、GC3)等一系列信息。
1.3.2 中性绘图分析 使用GC1与GC2的平均值(GC12)与GC3作为数据绘制中性对比图,中性对比图可以用来检测密码子突变压力和选择压力的平衡,从而揭示GC12和GC3的关系[28]。在密码子偏好中性对比中,每个离散点表示1个基因,若GC12与GC3为中性,则这些点应位于对角线上,若不为中性,这些点应出现在横坐标的平行线上[29]。
1.3.3 ENC-plot分析 有效密码子数分析(ENC-plot)用于分析密码子使用受到选择压力和突变压力的影响程度,根据各组基因密码子的GC3和有效密码子数(ENC),首先计算出预期ENC(预期ENC=GC3+2+29/[GC32+(1-GC3)2]),然后使用R语言绘制ENC-plot图,通过比较预期ENC与实际ENC得出突变压力和选择压力对密码子使用偏好性的影响程度[8]。
1.3.4 PR2-plot分析 奇偶校验分析(PR2-plot)用于展现突变压力与选择压力对于密码子使用的影响程度,分析密码子第三位碱基的A、T、C、G含量(分别为A3、T3、C3、G3),并分别以G3/(G3+C3)和A3/(A3+T3)为横坐标和纵坐标进行PR2-plot绘图,各个基因的密码子偏好性通过其与中心点的方向和矢量偏差表示,而图中中心点表示A=T和C=G,即此时基因的密码子使用无偏好性[30]。
1.3.5 最优密码子确定 最优密码子表示基因组中使用频率最高的密码子,以ENC为首选标准,将48条叶绿体基因组按照ENC进行排序,ENC最高的5个基因组归为高表达基因组,ENC最低的5个基因组为低表达基因组。将同时满足高频[RSCU(同义密码子相对使用度)>1]和高表达[ΔRSCU(同义密码子相对使用度之差)≥0.08]的密码子作为最优密码子。
1.3.6 对应性分析 使用CodonW 1.4.2基于RSCU对山楂属11个种进行对应性分析,将山楂属这11个种所共有的48个编码蛋白质的基因组按照基因功能分为5种类型,通过分析其变异情况得到影响其密码子偏好性的主要影响因素。
1.4 系统发育分析
基于山楂属11个种构建叶绿体全基因组系统发育树和CDS系统发育树。先将山楂属11个种植物叶绿体全基因组和CDS通过MAFFT v.7软件进行比对[31],比对结果通过trimAl[32]进行修饰,修改后的比对文件基于RAxMLv.8中的GTR+I+G模型,采用最大似然法进行系统发育分析[33],设置1 000次自展值重复。
2 结果与分析
2.1 叶绿体基因组结构
山楂属植物叶绿体基因组呈现标准的四分体结构,分别为大单拷贝区、小单拷贝区、反向重复区a和反向重复区b,叶绿体基因组全长为159 607~159 875 bp(图1)。LSC长度为87 601~87 874 bp,SSC长度为19 139~19 312 bp,单个反向重复区长度为26 347~26 385 bp。各个种的G+C含量为36.6%~36.7%,基因总数为127~132个,其中rRNA数量均为8个,tRNA数量除C. scabrifolia为36个外其余均为37个,编码蛋白质的基因数量为83~85个(表2)。综合来看,山楂属植物的叶绿体基因组G+C含量相近,基因种类和数量相近,未发现IR区丢失现象,叶绿体基因组长度变异较小,结构未发现明显差异。
2.1.1 重复序列分析 如图2A所示,在本研究中,单核苷酸、二核苷酸、四核苷酸和复合重复序列均在山楂属植物中被检测到,在本研究所选取的山楂属植物中,检测到的单核苷酸重复序列数量介于41~55,在各个种中单核苷酸重复序列数量均排第一位,而单核苷酸重复序列数量最多的物种为C. hupehensis,最少的物种为C. marshallii。山楂属植物中二核苷酸重复序列数量总体差异不大,C. oresbia被检测到的二核苷酸重复序列数量最少,为13个,C. maximowiczii、C. kansuensis、C. cuneata、和C. bretschneideri数量最多,为15个,其余物种则为14个。三核苷酸重复序列仅在C. hupehensis、C. cuneata、C. marshallii、C. pinnatifida和C. scabrifolia中被检测到,四核苷酸重复序列数量为3~5个,各物种之间差异不大。五核苷酸重复序列仅在C. marshallii中被检测到,六核苷酸重复序列仅在C. cuneata和C. marshallii中被检测到。这一结果说明山楂属植物的简单重复序列的类型和数量有部分相似之处,但总体来看也有一定的差异。
使用 REputer 在线工具对 11 种山楂属植物叶绿体基因组的离散重复序列进行分析,统计结果如图2B所示,结果显示山楂属植物离散重复序列具有一定相似性,回文重复序列为23~28,正向重复序列为20~29,其中C. kansuensis的回文重复序列与反向重复序列的数量均为最多,而C. marshallii的2种重复序列的数量均为最少。反向重复序列为3~11个,其中C. kansuensis的反向重复序列数量远高于其他10个种,为11个。互补重复序列在C. hupehensis中检测到的数量最多,为5个,而在C. maximowiczii与C. bretschneideri中并未检测出互补重复序列。总的来说山楂属植物的离散重复序列存在着一定的差别。
2.1.2 边界扩张收缩分析 对山楂属植物的边界扩张收缩分析结果(图3)表明,山楂属11个种植物的大单拷贝区与反向重复区b的边界(JLB)均位于rps19基因中,除C. marshallii和C. bretschneideri外,其余9個种的rps19基因均有120 bp位于IRb区域中;反向重复区b与小单拷贝区的边界(JSB)均位于ndhF中,且ndhF位于IRb的长度均为12 bp;JSA 均存在于ycf1基因中,且均有1 074 bp位于IRa中,rpl2为11个种植物的共有基因,均位于大单拷贝区与反向重复区a(JLA)的左侧,其中有9个种植物rpl2基因与JLA距离为190 bp,而C. marshallii和C. bretschneideri的rpl2基因与JLA的距离则发生了变异,与其余9个种植物略有不同。总的来说,山楂属植物的叶绿体基因组进化关系保守,结构差异较小,边界扩张收缩幅度较为稳定,只发生了较小的变异。
2.1.3 共线性分析 使用Mauve软件,采用多重基因组比较法对山楂属11个种植物的叶绿体基因组进行共线性分析,山楂属植物叶绿体基因组结构与各个基因的排列顺序基本一致,共线性良好,未发现倒置和重排现象,叶绿体基因组之间具有高度相似性。
2.2 密码子偏好性
2.2.1 密码子组成分析 在研究密码子的使用偏好性时,ENC常用于评价物种密码子偏好性的大小,其值为20~61,ENC值越大表示密码子的偏好性越弱。一般认为,ENC值在35以下时可表明其密码子偏性现象较为显著[34]。由表3可知,山楂属11个种植物的叶绿体基因组平均ENC为46.61~47.55,均大于35,密码子偏好性较弱,密码子总G+C含量与第一、第二、第三位的G+C含量均小于50%,且呈现出GC1>GC2>GC3的趋势,说明山楂属植物的叶绿体基因组富含A和T 2种碱基,且偏好于使用A、T作为密码子第三位结尾碱基。
2.2.2 PR2-plot绘图分析 若密码子的偏好性只受突变压力的影响,则A、T与C、G的使用频率应该是完全相等的。由图4可知,图中坐标点的分布并不均匀,可以明显看出,右侧的坐标点多于左侧,下方的坐标点多于上方,而分布于右下角区域的基因数量最多,说明山楂属植物叶绿体基因组密码子第三位碱基对于T的使用率大于A,对于G的使用率大于C,说明其密码子偏好性不只受到突变的影响,而是选择压力和突变压力共同作用的结果。
2.2.3 中性绘图分析 山楂属植物中性绘图分析见图5,各基因的GC3取值为20.74%~36.54%,GC12的值则介于31.75%~53.96%,回归系数为0.364~0.388,GC12与 GC3的相关系数为0.324~0.525,双尾检验均未达到显著水平(P>0.05),GC12与GC3之间相关性不显著,选择压力对其密码子的偏好性具有显著影响,说明山楂属植物叶绿体基因组密码子的第一、第二位碱基与第三位碱基的组成相关性较弱,密码子受选择压力的影响较大。
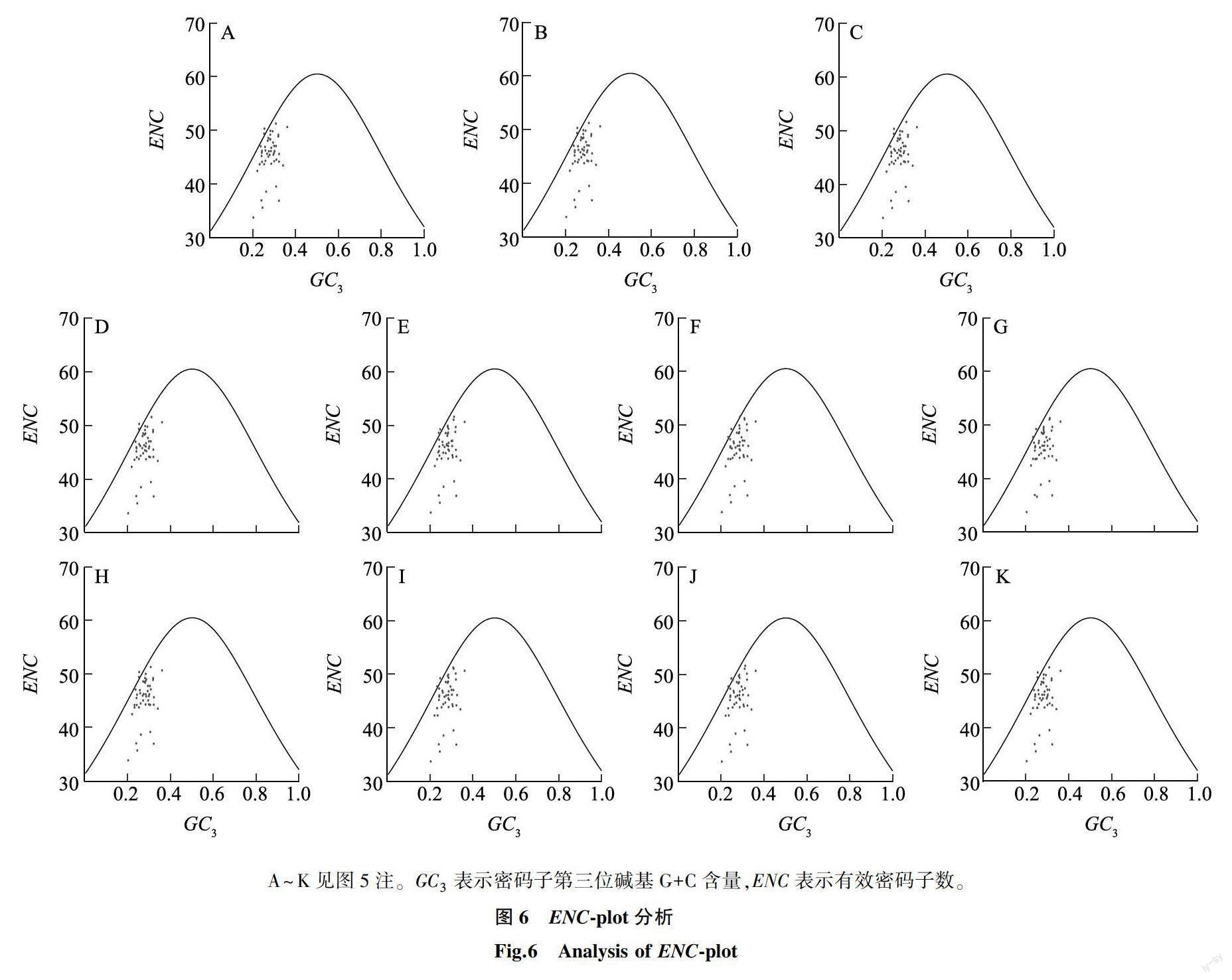
2.2.4 ENC-plot绘图分析 ENC-plot绘图能够揭示基因组密码子的ENC与 GC3之间的联系,如图6所示,坐标点大多分布在标准ENC曲线下方,且大多与预期ENC差距很大,即大部分基因的实际ENC小于预期值,这部分基因主要受到自然选择的影响。仅有少数基因靠近标准曲线,即只有少数基因的密码子偏好性主要受到突变压力的影响。总的来说,在本研究中,自然选择压力是供试山楂属植物叶绿体基因组密码子偏好性的主要影响因素。
2.2.5 山楂属植物最优密码子 对48个CDS基因按照ENC进行排序,根据高表达基因和低表达基因中密码子的RSCU和△RSCU来确定其最优密码子,筛选得到的最优密码子如表4所示,最优密码子数量介于17~20个,C. kansuensis、C. oresbia、C. pinnatifida的最优密码子数量最多,C. marshallii的最优密码子数量最少,分析它们的最优密码子数据可知,山楂属11个种植物的最优密码子都大多以A或U作为第三位碱基,说明其最优密码子偏向于使用A和U作为结尾。对其共有最优密码子进行分析,发现其共有最优密码子有13个,分别为GCA、GCU、AGA、CGA、UGU、CAA、UUA、UUU、AGU、UCU、ACA、UAU和GUU,其中有6个以A作为末碱基,7个以U作为末位碱基,共有密码子的第三位碱基均为A和U。差异密码子有7个,分别为GAC、GAA、GGA、AUA、CUU、AAA和ACC,存在差异的最优密码子中,有4个以A作为第三位碱基,2个以C作为末位碱基,1个以U作为末位碱基。分析山楂属11个种植物的最优密码子发现,不存在以G作为末位碱基的最优密码子。
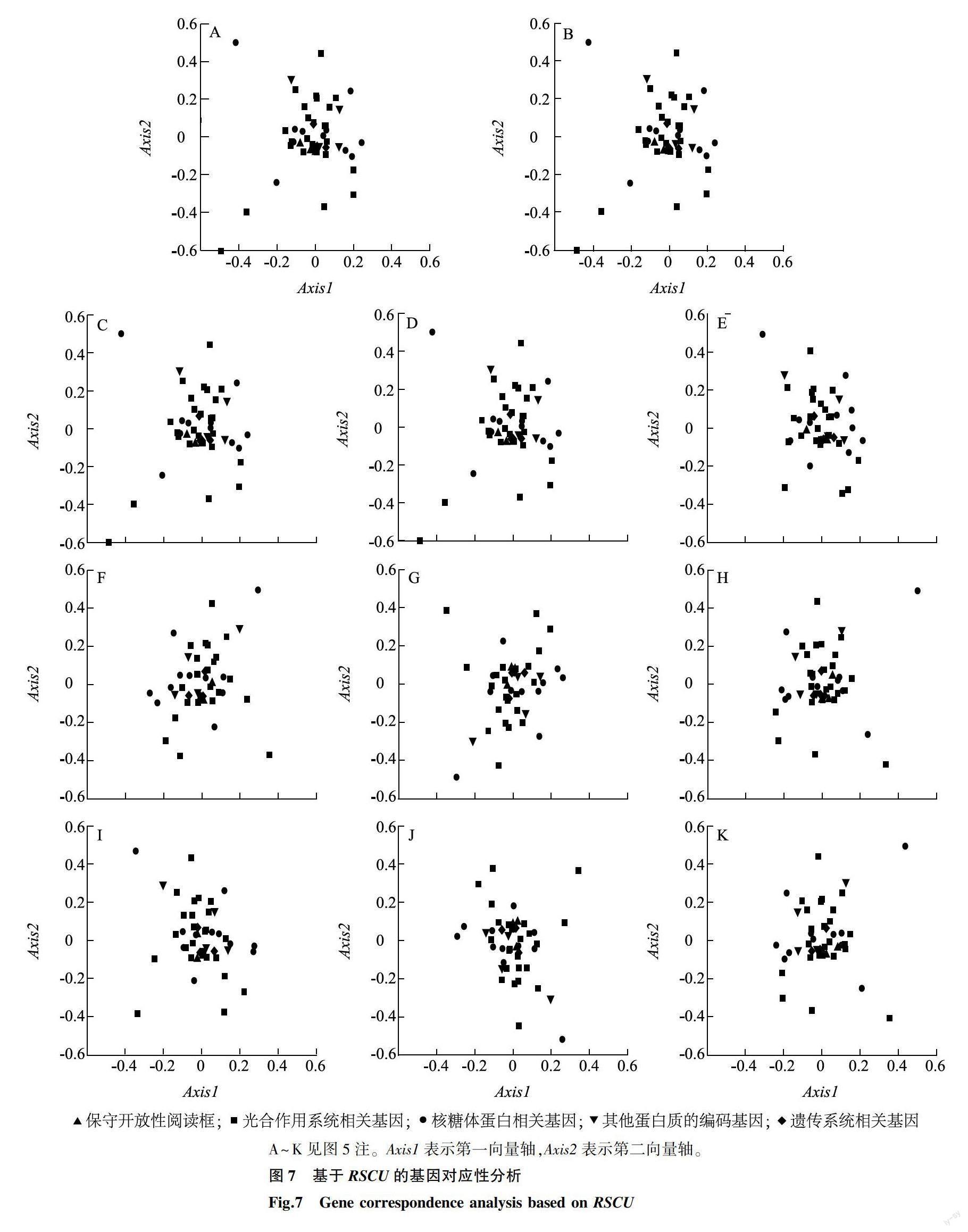
2.2.6 对应性分析 基于RSCU对山楂属植物叶绿体基因组48个共有CDS进行对应性分析,结果显示,其第一轴贡献率为11.69%~12.02%,第二轴贡献率为8.78%~8.94%,第三轴贡献率为8.22%~8.37%,第四轴贡献率为7.74%~8.02%,前四轴累计贡献率为36.71%~37.23%,第一轴对变异的贡献率与其他3个轴相差较大,为影响其变异的主要因素。为了深入分析其密码子偏好性特征,使用48个CDS的第一轴和第二轴建立平面坐标系,结果(图7)显示,山楂属11个种植物的CDS序列在平面中的分布相似性很高,均显示遗传系统相关基因与保守性开放阅读框的分布相对更加集中,说明这2类功能的基因内部存在相似的密码子使用偏好性。而其余3种功能的基因分布相对更加分散,说明这3种基因的密码子偏好性差异较大。
2.3 系统发育分析
对基于叶绿体CDS构建的系统进化树(图8A)与基于叶绿体全基因组构建的系统发育树(图8B)进行分析,结果显示,2种系统发育树具有很高的相似性,C. kansuensis、C. oresbia、C. chungtienensis、C. bretschneideri、C. maximowiczii、C. rhipidophylla和C. marshallii在2种系统进化树中具有相同的系统发育位置。但2种系统发育树也显现出了一定的差异,基于叶绿体CDS构建的系统发育树显示C. scabrifolia被单独归为一个远缘分支,显示其与另外10个种的亲缘关系较远;基于叶绿体全基因组序列构建的系统发育树(图8B)则将C. cuneata单独归为一个远缘分支。除此之外,基于叶绿体CDS构建的系统发育关系显示,C. hupehensis与C. pinnatifida亲缘关系密切,聚为一类,而基于叶绿体全基因组构建的系统发育树则为C. hupehensis、C. pinnatifida和C. scabrifolia聚為一支。总的来说,叶绿体基因组的2种系统发育树展现出来的系统发育关系既存在着部分差异,也存在着一定的相似性。
3 讨论与结论
植物叶绿体全基因组长度大多为120~200 kb,包含植物体丰富的遗传学信息[35]。随着第二代高通量测序技术的发展和测序成本的降低,已有大量的叶绿体基因组数据被上传至GenBank公共数据库,为植物的系统发育和分子标记研究提供了重要的参考。本研究对山楂属11个种的植物叶绿体基因组进行了系统发育与密码子偏好性分析,对于深入研究山楂属植物的进化关系具有一定的意义。本研究选取了山楂属11个种的植物叶绿体基因组进行分析,结果显示,山楂属植物的叶绿体基因组结构保守,叶绿体基因组长度变异较小,未发现任何基因组倒置和重排现象,这与悬钩子属植物叶绿体基因组的情况相似[36],但在樟科植物的研究中发现,IR区存在着部分基因重排现象[37],这与本研究的结果存在一定的差异。重复序列包含植物体的重要进化信息,是控制植物体生长发育的重要部分,重复序列的差异会对植物的遗传发育产生重要影响[38],对所选取的山楂属植物的离散重复序列进行分析,发现正向重复序列、回文重复序列、反向重复序列3种离散重复序列在山楂属11个种植物中均有分布,而互补重复序列在C. maximowiczii与C. bretschneideri中并未检测出,推断C. maximowiczii与C. bretschneideri在系统发育关系上可能存在着一定的相似性,这种推断与本研究中2种系统进化树展现的系统发育关系也相吻合。
分子进化中性理论认为,基因的碱基突变对密码子的影响是中性的或近似中性的[39]。但如果基因组的密码子受到外界环境选择的影响,则会导致密码子的使用和碱基组成出现偏向性[40]。本研究中选取的山楂属植物叶绿体基因组密码子的GC12与 GC3的相关系数为0.324~0.525,相关性均未达到显著水平(P>0.05),GC12与GC3之间相关性较弱,山楂属植物叶绿体基因组密码子的第一、第二位碱基与第3位碱基差异较大,说明选择压力对其密码子有着非常大的影响,而ENC-plot和PR2-plot绘图分析结果也表明,山楂属植物叶绿体基因组的密码子受选择压力的影响较大。综合以上分析可以看出,本研究中的山楂属植物密码子使用受自然选择因素的影响远大于碱基突变,而影响密码子使用偏好性的主要因素在不同植物物种中也可能存在差异。对应性分析结果显示,遗传系统相关基因与保守性开放阅读框2种功能的基因呈现出相似的密码子使用偏性,而其余3种功能基因的密码子偏好性存在较大差异,推测这3种功能基因的密码子偏好性可能受到多种因素的共同影响。另外,本研究在山楂属11个物种中筛选得到17~20个最优密码子,在这11个物种中,均以A、U作为结尾的最优密码子数量最多,这一结果与乌头属植物[41]和睡莲属植物[42]的情况相似。分析其共有密码子发现,其共有最优密码子有13个,且均以A和U作为结尾,所有物种中均未发现以G作为末位碱基的最优密码子。最优密码子的筛选结果可以为后续山楂属植物的遗传育种工作提供重要的参考依据。
基于CDS和叶绿体全基因组构建的2种系统发育树展现出来的系统发育关系具有相似性,这也说明了编码蛋白质氨基酸序列的碱基突变与生物的进化历程存在一定联系,基于叶绿体基因组CDS的系统发育关系能在一定程度上对物种的系统发育关系和生物进化历程进行补充。但C. cuneata、C. hupehensis、C. pinnatifida和C. scabrifolia在2种系统发育树中的位置存在一定的差别,推测可能是其存在较为特殊的生物进化历程或非编码区碱基序列存在较大差异所导致的。此外,本研究也对山楂属植物的简单重复序列进行了鉴定和分析,可以为后续山楂属植物的分子標记研究提供一定的参考。总之,本研究对山楂属11个种植物的叶绿体基因组特征、密码子偏好性及系统发育关系进行了分析,对后续山楂属植物密码子优化、基因组改造以及探索其系统进化关系和增加外源基因表达量具有重要的参考价值。
本研究使用生物信息学手段,对山楂属植物叶绿体基因组进行分析,发现山楂属植物叶绿体基因组结构保守,未发现基因倒置和重排现象,边界扩张收缩幅度小,长度变异保守。对其简单重复序列与离散重复序列进行鉴定,重复序列的种类和数量存在一定的差异。对其密码子偏好性进行分析,结果显示,选择压力均对其密码子偏好性产生深刻的影响,筛选得到的最优密码子数量为17~20个,使用山楂属11个种植物的叶绿体全基因组和CDS分别构建系统发育树,发现这2种山楂属系统发育树展现出的系统发育关系存在一定相似性。
参考文献:
[1] 费开伟.读山楂种质资源专著——《中国果树志·山楂卷》[J].园艺学报,1998 (1):103.
[2] DEKIC V, RISTIC N, DEKIC B, et al. Phenolic and flavonoid content and antioxidant evaluation of hawthorn (Crataegus monogyna Jacq.) fruits and leaves extracts[J]. Bulletin of Natural Sciences Research, 2020, 10(1): 20-25.
[3] LISTON A, WEITEMIER K A, LETELIER L, et al. Phylogeny of Crataegus (Rosaceae) based on 257 nuclear loci and chloroplast genomes: evaluating the impact of hybridization[J]. PeerJ, 2021, 9: e12418.
[4] CHEN X L, ZHOU J G, CUI Y X, et al. Identification of Ligularia herbs using the complete chloroplast genome as a super-barcode[J]. Frontiers in Pharmacology, 2021, 9: 695.
[5] LI J, LI H Y, ZHI J K, et al. Codon usage of expansin genes in Populus trichocarpa[J]. Current Bioinformatics, 2017, 12(5): 452-461.
[6] MORALES-BRIONES D F, KADEREIT G, TEFARIKIS D T, et al. Disentangling sources of gene tree discordance in phylogenomic data sets: testing ancient hybridizations in Amaranthaceae sl[J]. Systematic Biology, 2021, 70(2): 219-235.
[7] CHAKRABORTY S, YENGKHOM S, UDDIN A. Analysis of codon usage bias of chloroplast genes in Oryza species[J]. Planta, 2020, 252(4): 1-20.
[8] TANG D F, WEI F, CAI Z Q, et al. Analysis of codon usage bias and evolution in the chloroplast genome of Mesona chinensis Benth[J]. Development Genes and Evolution, 2021, 231(1): 1-9.
[9] 王存堂,李子钰,张福娟,等.山楂属果实不同组织乙醇提取物的抗氧化成分及性能研究[J].食品与发酵工业,2021,47(16):117-122.
[10]AIERKEN A, BUCHHOLZ T, CHEN C, et al. Hypoglycemic effect of hawthorn in type II diabetes mellitus rat model[J]. Journal of the Science of Food and Agriculture, 2017, 97(13), 4557-4561.
[11]MIN Q, BAI Y T, ZHANG Y C, et al. Hawthorn leaf flavonoids protect against diabetes-induced cardiomyopathy in rats via PKC-α signaling pathway[J]. Evidence-Based Complementary and Alternative Medicine, 2017. https://doi.org/10.1155/2017/2071952.
[12]张浣悠,邓秩童,黄嘉泳,等.山楂黄酮的保健功效及提取工艺研究进展[J].食品研究与开发,2021,42(12):212-217.
[13]张 枭,杜 潇,孙馨宇,等.利用SSR标记构建部分山楂资源的基因身份证[J].沈阳农业大学学报,2021,52(2):153-159.
[14]WU X E, LUO D L, ZHANG Y M, et al. Comparative genomic and phylogenetic analysis of chloroplast genomes of hawthorn (Crataegus spp.) in southwest China[J]. Frontiers in Genetics, 2022, 13. https://doi.org/10.3389%2Ffgene.2022.900357.
[15]WU L W, CUI Y X, WANG Q, et al. Identification and phylogenetic analysis of five Crataegus species (Rosaceae) based on complete chloroplast genomes[J]. Planta, 2021, 254(1): 1-12.
[16]TAI T H, TANKSLEY S D. A rapid and inexpensive method for isolation of total DNA from dehydrated plant tissue[J]. Plant Molecular Biology Reporter, 1990, 8(4): 297-303.
[17]JIN J J, YU W B, YANG J B, et al. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes[J]. Genome Biology, 2020, 21(1): 1-31.
[18]WICK R R, SCHULTZ M B, ZOBEL J, et al. Bandage: interactive visualization of de novo genome assemblies[J]. Bioinformatics, 2015, 31(20): 3350-3352.
[19]SHI L C, CHEN H M, JIANG M, et al. CPGAVAS2, an integrated plastome sequence annotator and analyzer[J]. Nucleic acids research, 2019, 47(W1): 65-73.
[20]KEARSE M, MOIR R, WILSON A, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data[J]. Bioinformatics, 2012, 28(12): 1647-1649.
[21]王 瑞,歐金梅,李 昕,等.基于简单重复序列标记的药用梅品种的身份证构建[J].安徽中医药大学学报,2020,39(6):62-67.
[22]BEIER S, THIEL T, MüNCH T, et al. MISA-web: a web server for microsatellite prediction[J]. Bioinformatics, 2017, 33(16): 2583-2585.
[23]KURTZ S, SCHLEIERMACHER C. REPuter: fast computation of maximal repeats in complete genomes[J]. Bioinformatics (Oxford, England), 1999, 15(5): 426-427.
[24]XIA E H, TONG W, WU Q, et al. Tea plant genomics: achievements, challenges and perspectives[J]. Horticulture research, 2020, 7. https://doi.org/10.1038/s41438-019-0225-4.
[25]梁凤萍,文祥宁,高赫一,等.菊科植物叶绿体基因组特征分析[J].基因组学与应用生物学,2018,37(12):5437-5447.
[26]DARLING A C, MAU B, BLATTNER F R, et al. Mauve: multiple alignment of conserved genomic sequence with rearrangements[J]. Genome Research, 2004, 14(7): 1394-1403.
[27]WALDVOGEL A M, PFENNINGER M. Temperature dependence of spontaneous mutation rates[J]. Genome Research, 2021, 31(9): 1582-1589.
[28]DE OLIVEIRA J L, MORALES A C, Hurst L D, et al. Inferring adaptive codon preference to understand sources of selection shaping codon usage bias[J]. Molecular Biology and Evolution, 2021,38(8):3247-3266.
[29]XING Y P, XU L, CHEN S Y, et al. Comparative analysis of complete chloroplast genomes sequences of Arctium lappa and A.tomentosum[J]. Biologia Plantarum, 2019,63(1): 565-574.
[30]DUAN H R, ZHANG Q, WANG C M, et al. Analysis of codon usage patterns of the chloroplast genome in Delphinium grandiflorum L. reveals a preference for AT-ending codons as a result of major selection constraints[J]. PeerJ, 2021, 9:e10787.
[31]KATOH K, STANDLEY D M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability[J]. Molecular Biology and Evolution, 2013, 30(4): 772-780.
[32]CAPELLA-GUTI?RREZ S, SILLA-MARTíNEZ J M, GABALDóN T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses[J]. Bioinformatics, 2009,25(15):1972-1973.
[33]LI W, ZHANG C P, GUO X, et al. Complete chloroplast genome of Camellia japonica genome structures, comparative and phylogenetic analysis[J]. PLoS One, 2019, 14(5): e0216645.
[34]MENSAH R A, SUN X, CHENG C, et al. Analysis of codon usage pattern of banana basic secretory protease gene[J]. Plant Diseases and Pests, 2019, 10(1): 1-9.
[35]童一涵,郑 倩,杜新明,等. 多齿红山茶叶绿体基因组序列特征分析[J]. 植物资源与环境学报,2022,31(5):27-36.
[36]于丽平,孙孟涛,贺志敏,等. 川莓和峨眉悬钩子叶绿体比较基因组学及其系统发育关系分析[J].分子植物育种, 2022. http://kns.cnki.net/kcms/detail/46.1068.S.20220729.1007.004.html.
[37]田永靖. 樟科植物比较叶绿体基因组与系统发育研究[D]. 南京: 南京大学, 2021.
[38]KELLER J, ROUSSEAU-GUEUTIN M, MARTIN G E, et al. The evolutionary fate of the chloroplast and nuclear rps16 genes as revealed through the sequencing and comparative analyses of four novel legume chloroplast genomes from Lupinus[J]. DNA Research, 2017, 24(4): 34.
[39]ZHANG R Z, ZHANG L, WANG W, et al. Differences in codon usage bias between photosynthesis-related genes and genetic system-related genes of chloroplast genomes in cultivated and wild solanum species[J]. International Journal of Molecular Sciences, 2018, 19(10): 3142.
[40]LIU H B, LU Y Z, LAN B L, et al. Codon usage by chloroplast gene is bias in Hemiptelea davidii[J]. Journal of Genetics, 2020, 99(1): 1-11.
[41]樊东昌,穆赢通,贾俊英,等. 烏头属药用植物叶绿体基因组密码子特征和系统发育分析[J].分子植物育种, 2022. http://kns.cnki.net/kcms/detail/46.1068.S.20220711.1339.002.html.
[42]毛立彦,黄秋伟,龙凌云,等. 7种睡莲属植物叶绿体基因组密码子偏好性分析[J].西北林学院学报,2022,37(2):98-107.
(责任编辑:陈海霞)
