DNA甲基化与非大动脉粥样硬化型卒中相关性研究进展
2023-06-07姜晓晴瓮佳旭周宏宇李子孝王拥军
姜晓晴,瓮佳旭,周宏宇,李子孝,3,4,王拥军,3,4
缺血性卒中是一种复杂且具有很强异质性的疾病,由多种环境因素和遗传风险因素共同决定[1]。目前临床多使用TOAST分型将缺血性卒中分为大动脉粥样硬化(largeartery atherosclerosis,LAA)、心源性栓塞(cardioembolism,CE)、小动脉闭塞(smallvessel occlusion,SVO)、其他明确病因(other determined etiology,ODE)和不明原因(undetermined etiology,UE)5种类型[2]。卒中的遗传背景相关风险为37.9%,而迄今发现的变异相关遗传风险仅占总遗传背景相关风险的5%~10%[3-4]。有更多卒中相关基因和可遗传危险因素尚未被发现,其中,部分可遗传危险因素可能与表观遗传学修饰有关。
表观遗传学修饰是环境因素与基因组的动态相互作用,在不改变DNA序列的基础上影响基因的表达或细胞表型[5]。DNA甲基化指发生在胞嘧啶-磷酸-鸟嘌呤(cytosinephosphate-guanine,CpG)二核苷酸中胞嘧啶上第5位碳原子的甲基化,是与基因组稳定性、基因表达和调控相关的最重要的表观遗传学机制。CpG岛为GC含量>55%且至少含500个碱基对的DNA片段,位于基因启动子区域的CpG岛甲基化可通过阻止转录因子结合等机制抑制基因表达[6]。卒中表观遗传学研究表明,缺血性卒中患者全基因组具有低甲基化的趋势,提示相关基因启动子可发生低甲基化,导致相关基因转录水平增加[7]。一项旨在检测与缺血性卒中发生和卒中亚型相关差异甲基化位点(differentially methylated positions,DMPs)的研究发现,不同卒中亚型存在不同程度的DNA甲基化差异[8]。DNA甲基化可能作为缺血性卒中及其亚型的潜在生物标志物。本文主要聚焦除LAA外其他类型卒中(CE、SVO、ODE)DNA甲基化相关的研究,探究特定候选基因的DNA甲基化水平对这些类型卒中病理生理的潜在影响,为缺血性卒中的诊断和治疗提供新思路。
1 DNA甲基化与心源性栓塞型卒中
1.1 心源性栓塞型卒中与雌激素受体α基因甲基化 既往有动物研究发现,在永久大脑中动脉闭塞(middle cerebral artery occlusion,MCAO)模型小鼠和大鼠中,雌激素受体α(estrogen receptor α,ERα)基因呈低甲基化[9]。Lin等[10]定量测量了201例缺血性卒中患者和217例健康对照的ERα启动子区域14个CpG位点的甲基化水平,发现LAA和CE亚型卒中有5个CpG位点呈现低甲基化水平。
ERα是雌激素发挥生物效应的主要核受体之一。多项研究提示,当卒中模型动物发生脑缺血损伤后,雌激素具有减轻神经毒性、抑制神经炎症反应等潜在作用,而雌激素发挥作用主要与局部区域ERα的表达增加有关[9,11-13]。Westberry等[14]的研究进一步提示,ERα基因甲基化是脑缺血损伤后ERα上调的主要机制之一。CE型卒中后特异性ERα低甲基化水平改变可能通过增加ERα的表达来介导雌激素的神经保护作用[10]。
1.2 心源性栓塞型卒中与ZFHX3基因甲基化Cullell等[15]研究发现,缺血性卒中患者与健康对照存在957个DMPs,其中锌指同源框3(zinc finger homeobox 3,ZFHX3)基因低甲基化与CE型卒中相关。研究者进一步通过孟德尔随机化分析发现,ZFHX3基因cg07786668与CE型卒中的发病风险具有因果关系[15]。ZFHX3基因编码转录因子AT模序结合因子1(AT-motif binding factor 1,Atbf1),而Atbf1可调节肌源性和神经元分化[16]。既往全基因组关联研究在基因水平上发现ZFHX3基因遗传变异与CE型卒中的发病风险增加相关[17-18]。Cullell等[15]的研究结果从表观遗传学水平为探究ZFHX3基因与CE型卒中的相关性提供了新的证据。
2 DNA甲基化与小动脉闭塞型卒中
2.1 小动脉闭塞型卒中与MMP-2基因甲基化 在缺血性卒中亚型中,仅在SVO型中发现MMP-2活性升高,且MMP-2基因单核苷酸多态性是SVO型卒中的独立危险因素[19]。Lin等[20]研究发现,与健康对照相比,SVO患者MMP-2启动子甲基化水平降低。MMP-2主要介导细胞外基质降解重塑,在卒中早期损伤中可通过降解基膜干扰血-脑脊液屏障功能,进一步参与卒中后出血转化和神经元凋亡的过程[21]。Arba等[22]研究发现血-脑脊液屏障功能失调是SVO型卒中的潜在发病机制之一,推测MMP-2启动子低甲基化导致MMP-2表达增加,过表达的MMP-2通过破坏血-脑脊液屏障,参与SVO型卒中的病理生理学过程。
2.2 小动脉闭塞型卒中与CYP26C1基因甲基化 Lee等[23]研究发现SVO型卒中患者细胞色素P450 26C1(cytochrome P450 family 26 subfamily C member 1,CYP26C1)启动子甲基化水平明显降低。CYP26C1是细胞色素P450家族成员,参与视黄酸的分解代谢,可催化视黄酸分解为羟基视黄酸[24]。目前相关动物实验提示,在小鼠和大鼠MCAO模型中应用视黄酸具有减轻神经炎症、促进运动功能恢复的作用[25-26]。临床研究表明,在卒中患者中可观察到9-顺式视黄酸水平显著降低,而高血浆视黄酸水平可以延缓高血压患者首次缺血性卒中的发生[27-28]。CYP26C1可能通过视黄酸代谢与卒中发病建立潜在联系。此外,既往研究发现视黄酸可以降低血浆Hcy水平[29],而高同型半胱氨酸血症为SVO型卒中的重要危险因素之一[30],推测CYP26C1低甲基化后自身表达增加,可能通过促进视黄酸分解,降低血视黄酸水平,进而影响Hcy水平,从而参与SVO的发生。
3 DNA甲基化与其他明确病因型卒中
ODE型卒中主要涉及血管因素和血液因素两方面的疾病。血管因素包括各种原因引起的血管炎、血管畸形(如动-静脉畸形、烟雾病)等。血液因素包括骨髓增殖性肿瘤(红细胞增多症、血小板增高)、镰状细胞病、高凝状态(如抗磷脂抗体综合征、系统性红斑狼疮等导致)等[31]。
3.1 DNA甲基化与系统性血管炎
3.1.1 抗中性粒细胞胞质抗体相关性血管炎 抗中性粒细胞胞质抗体相关性血管炎(antineutrophil cytoplasmic antibodyassociated vasculitis,AAV)表现为全身小血管坏死性炎症,主要发病机制为机体产生针对自身中性粒细胞胞质蛋白的抗体,即抗髓过氧化物酶(myeloperoxidase,MPO)和抗蛋白酶3(proteinase 3,PRTN3)抗体,导致中性粒细胞过度激活,产生活性氧及中性粒细胞胞外陷阱[32]。正常中性粒细胞主要在细胞发育的早期表达MPO和PRTN3,但AAV患者的中性粒细胞从早期至成熟均持续表达这两种蛋白,提示MPO和PRTN3两种基因沉默发生异常,介导基因沉默的DNA甲基化可能在其中发挥作用[33]。
Jones等[34]研究发现,与健康对照及非活动期AAV患者相比,活动期AAV患者的白细胞MPO(外显子5-6的CpG岛)和PRTN3(启动子)基因呈低甲基化状态。纵向收集AAV患者样本,发现疾病缓解期PRTN3启动子甲基化水平存在升高和降低两种变化,PRTN3启动子甲基化水平降低的患者卒中复发风险是升高患者的4.55倍,提示PRTN3启动子甲基化水平的变化可能具有预测AAV复发的价值。
在AAV 患者中,除观察到MPO和PRTN3甲基化水平改变,Ciavatta等[35]还发现AAV患者粒性白细胞中人类runt相关转录因子3(runt-related transcription factor 3,RUNX3)基因启动子呈高甲基化,同时发现组蛋白H3第27位赖氨酸的三甲基化修饰(histone-3 lysine-27 trimethylation,H3K27me3)标记缺失。H3K27me3是一类重要的转录抑制性翻译后修饰,参与组蛋白H2A泛素化修饰等机制,促进染色质凝集,介导基因沉默[36]。RUNX3与多梳抑制复合物2(polycomb repressive complex 2,PRC2)亚单位相互作用,参与催化并维持H3K27me3[35]。基于以上发现,Ciavatta等[35]提出了调控模型理论:RUNX3高甲基化引起自身表达降低,导致H3K27me3来源缺失,同时组蛋白H3K27me3去甲基化酶(jumonji domaincontaining protein 3,JMJD3)不断动态去除H3K27me3标记,使MPO和PRTN3基因沉默异常,增加染色质可及性,提高两种基因的转录水平。
3.1.2 巨细胞动脉炎 巨细胞动脉炎(giantcell arteritis,GCA)主要累及大动脉和中动脉,以形成肉芽肿为主要特征,可导致血管闭塞从而引发卒中[37]。Coit等[38]发现GCA患者颞动脉样本中T细胞相关基因特异性甲基化水平改变,包括蛋白磷酸酶3催化亚基γ同工酶(protein phosphatase 3,catalytic subunit,gamma isozyme,PPP3CC)、活化T细胞核因子2(nuclear factor of activated T cell 2,NFATC2)、活化T细胞核因子1(nuclear factor of activated T cell 1,NFATC1)基因相关位点呈低甲基化。上述3个基因主要参与T细胞受体(T cell receptor,TCR)/CD28信号和钙调神经磷酸酶(calcineurin,CaN)/T细胞活化核因子(nuclear factor of activated T cell,NFAT)通路。NFAT是调节干扰素γ(interferon-gamma,IFNG)、TNF及CD40配体(CD40LG)等细胞因子表达的转录因子,其基因低甲基化可导致T细胞激活和分化活动增强。该研究同时发现IFNG、IL-6、IL-21、TNF、趋化因子受体7(chemokine receptor 7,CCR7)、趋化因子配体18(chemokine ligand 18,CCL18)等多种促炎因子基因呈低甲基化,可导致血管壁细胞因子的炎性浸润,产生疾病促炎环境。T细胞激活和分化、细胞因子炎性浸润是GCA发生、发展的重要环节,上述基因低甲基化水平改变在GCA发病中具有潜在作用。
除局部炎性细胞浸润形成肉芽肿所致组织损伤外,IL-6驱动下的系统性炎症同样为GCA发病机制的重要环节。单核细胞是系统性炎症的主要参与因素[39]。Estupiñán-Moreno等[40]对82例GCA患者和31例健康对照者的CD14+单核细胞进行了全表观基因组关联分析,发现与健康对照相比,GCA患者CD14+单核细胞在1190个基因上存在1371个DMPs,其中85%以上的DMPs呈高甲基化水平改变。基因本体学分析发现,上述DMPs呈现免疫反应功能通路(如调节白细胞趋化性、IFNG和整合素生成)及单核细胞生物学通路(如巨噬细胞分化和增殖、巨噬细胞集落刺激因子等细胞因子产生)的显著富集。进一步分析发现,与经糖皮质激素治疗后疾病缓解的GCA患者相比,疾病活动期GCA患者的CD14+单核细胞存在688个DMPs,其中85%以上的DMPs呈低甲基化水平改变,主要呈现GCA免疫致病过程相关通路的富集(如对IL-6、IL-11的细胞应答)。上述研究结果提示,DNA甲基化在GCA发病、疾病活动性和激素治疗反应方面发挥潜在作用,可能作为可识别的生物标志物,有助于GCA疾病的早期诊断、分类和治疗。
3.1.3 川崎病 川崎病(Kawasaki disease,KD)是一种高热性血管炎疾病,累及多个系统脏器和组织,尤其是中小动脉[41]。Fcγ受体Ⅱa(FcγRⅡa,FCGR2A)基因存在的特殊遗传变异位点,可作为川崎病的潜在风险标志[42]。Kuo等[43]研究发现KD患者全血细胞FCGR2A启动子区域发生低甲基化。Li等[44]的研究发现,经过静脉注射免疫球蛋白治疗后,KD患者的FCGR2A甲基化水平可恢复正常。FCGR2A蛋白在巨噬细胞、中性粒细胞、单核细胞和树突细胞表面表达,可增加吞噬作用和炎性介质的产生[45]。另外,Huang等[46]研究发现在KD患者全血细胞中,Toll样受体(Toll-like receptor,TLR)基因同样发生低甲基化,并且同样经治疗后可恢复至正常甲基化水平。TLR可与FCGR2A相互作用,诱导TNF-α等炎性介质产生[47]。上述研究提示FCGR2A与TLR基因发生低甲基化可能导致促炎反应增强,参与KD的发生发展。
3.1.4 白塞病 白塞病(Behcet’s disease,BD)血管炎多侵犯小动脉、小静脉,以侵犯全身极微小的微血管为主,可导致血管坏死或破裂、管腔狭窄、血栓形成及血管动脉瘤样改变,从而造成器官或组织的损害[48]。Hughes等[49]发现,与健康对照相比,BD患者外周血CD4+T细胞和单核细胞中与细胞骨架重塑相关的基因呈现甲基化水平改变:肌球蛋白基因如肌球蛋白重链15(myosin heavy chain 15,MYH15)、肌球蛋白1D(myosin 1D,MYO1D)、肌球蛋白磷酸酶Rho相互作用蛋白(myosin phosphatase Rho interacting protein,MPRIP)基因呈高甲基化,肌球蛋白1C(myosin 1C,MYO1C)基因呈低甲基化;肌动蛋白相关基因如脑特异性血管生成抑制因子1相关蛋白2样蛋白1(brain-specific angiogenesis inhibitor 1-associated protein 2-Like protein 1,BAIAP2L1)、锚蛋白1(ankyrin 1,ANK1)基因呈高甲基化;Ras相关的C3肉毒素底物1(ras-related C3 botulinum toxin substrate 1,RAC1)、肌动蛋白束蛋白2(fascin-2,FSCN2)、丫叉同源物1(slingshot homolog 1,SSH1)基因呈低甲基化。经秋水仙碱治疗后,BD患者特定基因甲基化水平可恢复正常。经治疗后的BD患者与治疗前相比,介导微管形成与组织的驱动蛋白家族成员2A(kinesin family member 2A,KIF2A)呈高甲基化改变,促微管聚合蛋白(tubulin polymerization promoting protein,TPPP)呈低甲基化改变。这些参与细胞骨架动力学和细胞迁移的基因甲基化水平变化有作为BD潜在生物标志物与治疗靶点的可能。
3.2 DNA甲基化与血管畸形
3.2.1 动静脉畸形 在脑动静脉畸形(brain arteriovenous malformations,BAVM)患者中,磷脂酶A2 Ⅶ型(phospholipase A2 group Ⅶ,PLA2G7)、细胞周期蛋白依赖的激酶抑制剂2A(cyclin-dependent kinase inhibitor 2A,CDKN2A)、一氧化氮合成酶1接头蛋白(nitric oxide synthase 1 adaptor protein,NOS1AP)、血小板衍生生长因子D(platelet derived growth factor D,PDGFD)基因特定CpG岛区域呈现高甲基化水平[50-53]。上述基因甲基化水平改变可作为BAVM的预测因素,其具体机制可能涉及脂质代谢、内皮细胞增殖、血管发育、血管损伤修复等途径。上述研究为探究BAVM发病机制提供了新思路,其结果提示应关注参与以上过程的重要基因甲基化水平改变。
3.2.2 烟雾病 Sung等[54]研究发现烟雾病(moyamoya disease,MMD)患者内皮集落形成细胞中分拣蛋白1(sortilin 1,SORT1)基因启动子呈低甲基化,且对MMD具有一定的诊断价值。SORT1作为VPS10P结构域受体家族的一员,参与胞内蛋白向细胞膜或膜性细胞器的运输,介导细胞内吞和溶酶体降解作用[55]。SORT1基因低甲基化可使SORT1过度表达,导致促血管生成因子及MMP-9表达增加,血管生成抑制因子血小板反应蛋白2(thrombospondin 2,THBS2)表达下降。由于血管生成本身是促进和抑制生成两种因素的动态相互作用,推测SORT1基因低甲基化水平改变是通过调节生成、抑制因子的表达水平,破坏血管生成过程的动态平衡,从而对MMD发病产生影响。
3.3 DNA甲基化与骨髓增殖性肿瘤 骨髓增殖性肿瘤(myeloproliferative neoplasm,MPN)包括真性红细胞增多症(polycythemia vera,PV)、原发性血小板增多症(essential thromboc y themia,E T)、骨髓纤维化(myelofibrosis,MF)3种类型[56],目前已开展的研究集中在PV和ET这两种疾病类型与DNA甲基化的相关性方面。
3.3.1 骨髓增殖性肿瘤体细胞突变 MPN存在Janus激酶2(Janus kinase 2,JAK2)基因的特异性驱动突变,是驱动疾病发生的主要原因之一[57]。JAK2的主要突变形式为JAK2V617F,可在95%的PV和50%~60%的ET患者中检测发现[58]。相关研究发现,参与DNA甲基化过程的4种基因发生体细胞突变,在MPN发病过程中发挥作用,这4种基因分别为TET甲基胞嘧啶双加氧酶2(TET methylcytosine dioxygenase 2,TET2)、DNA甲基转移酶3A(DNA methyltransferase 3A,DNMT3A)、异柠檬酸脱氢酶1(isocitrate dehydrogenase 1,IDH1)和异柠檬酸脱氢酶2(isocitrate dehydrogenase 2,IDH2)基因[59-61]。
TET2突变为功能缺失性,在MPN中占12%~17%[62]。TET2蛋白催化DNA中5-甲基胞嘧啶转化为5-羟甲基胞嘧啶,介导DNA去甲基化。TET2突变多与JAK2V617F突变同时发生,且两种突变顺序可影响疾病表型,先发生JAK2突变在PV患者中多见,先发生TET2突变在ET患者中多见[59]。
DNMT3A作为DNA甲基化转移酶家族的一员,参与DNA从头甲基化过程。DNMT3A基因发生移码突变或无义突变,导致甲基转移酶活性降低。与TET2基因类似,PV患者先发生JAK2突变可能性大,而ET患者先发生DNMT3A突变可能性大[60]。
IDH1和IDH2基因编码两种表观遗传调控因子,参与DNA甲基化和组蛋白修饰过程。IDH1和IDH2蛋白催化异柠檬酸氧化脱羧生成α-酮戊二酸。IDH1和IDH2残基发生点突变,将α-酮戊二酸转化为2-羟基戊二酸,通过表观遗传调控相关基因,从而促进白血病发生,在介导MPN转化为急性白血病的过程中发挥潜在作用[61]。
3.3.2 骨髓增殖性肿瘤与核因子-κB信号通路基因甲基化 有研究在PV和ET患者的骨髓和外周血中发现特定基因呈低甲基化,分别为C型凝集素域家族7成员A(C-type lectindomain family 7,member A,CLEC7A)和外异蛋白A受体(ectodysplasin A receptor,EDAR)关联死亡域(EDARassociated death domain,EDARADD)基因,这两种基因主要参与核因子-κB(nuclear factor kappa binding,NF-κB)信号通路[63]。NF-κB介导炎症诱发的癌变和骨髓增殖性疾病的进展[64],因此推测上述低甲基化水平基因对MPN发病机制具有潜在影响。
3.4 DNA甲基化与高凝状态 高凝状态指遗传性或后天获得性血栓好发倾向,分为原发性与继发性高凝状态。抗磷脂抗体综合征和系统性红斑狼疮作为自身免疫性疾病,是导致继发性高凝状态的重要病因。
3.4.1 抗磷脂抗体综合征 抗磷脂抗体综合征(antiphospholipid syndrome,APS)以反复血栓形成、妊娠并发症为临床特征,伴血清自身抗体的升高[65]。
Weeding等[66]研究发现APS患者中性粒细胞中V-Ets骨髓成红细胞增多症病毒E26 癌基因同源物1(V-Ets erythroblastosis virus E26 oncogene homolog 1,ETS1)、表皮膜蛋白2(epithelial membrane protein 2,EMP2)、催产素(oxytocin,OXT)基因呈低甲基化,这些基因主要与妊娠相关。ETS1编码转录因子参与干细胞发育和细胞衰老,为系统性红斑狼疮的易感基因[67]。研究者同时发现蛋白酪氨酸磷酸酶非受体型2(protein tyrosine phosphatase,non-receptor type 2,PTPN2)基因呈低甲基化。PTPN2负向调节T细胞激活,PTPN2基因变异与类风湿性关节炎、炎性肠病等自身免疫性疾病风险增加有关[68-69]。
Patsouras等[70]研究发现在APS患者全血细胞中,IL-8启动子甲基化水平降低,组织因子(tissue factor,F3)基因体甲基化水平升高。有研究者体外使用抗磷脂抗体复合物刺激健康对照组单核细胞,发现IL-8启动子和F3基因体甲基化水平先升高后降低,最终导致基因过度表达。这一过程伴随甲基化CpG结合蛋白-2(methyl-CpG-binding protein 2,MECP2)、DNMT3B、TET1、组蛋白去乙酰化酶9(histone deacetylase 9,HDAC9)、AT相互作用功能域5B(AT-rich interactive domain 5B,ARID5B)等表观遗传修饰酶表达增加。提示在表观遗传修饰酶作用下,IL-8与F3甲基化水平变化可能参与APS的发病过程。
3.4.2 系统性红斑狼疮 系统性红斑狼疮(systemic lupus erythematosus,SLE)是一种影响多个器官的全身性自身免疫性疾病,其特征是针对细胞和组织产生异常免疫反应,形成自身抗体和免疫复合物沉积,进而导致炎症和器官损伤[71]。
Richardson等[72]研究发现,活动期SLE患者的CD4+T细胞中全基因组DNA甲基化水平降低,且与T细胞自身反应性相关,提示DNA甲基化水平异常与SLE的发生有关。Corvetta等[73]进一步研究发现,DNA甲基化抑制剂5-氮杂胞苷与SLE患者的疾病活动性有关,使用DNA甲基化抑制剂处理健康对照组的CD4+T细胞可诱导狼疮样综合征[74-75]。
目前研究发现多种IL基因甲基化水平改变与SLE相关。Lin等[76]发现SLE患者白细胞中IL-10和白细胞介素1受体2(type 2 IL-1 receptor,IL-1R2)基因启动子甲基化水平降低,且这两种基因低甲基化程度与SLE活动性呈正相关。Mi等[77]研究发现SLE患者外周血单个核细胞中IL-6基因启动子呈低甲基化,后续研究发现IL-6启动子低甲基化程度与SLE患者的肾脏损害程度呈正相关,与血浆补体C3、C4水平呈负相关[78]。IL在免疫反应中具有传递信息,以及激活与调节免疫细胞的作用,IL-10、IL-1R2、IL-6启动子低甲基化水平可作为临床诊断SLE的新型生物标志物。
Zhao等[79]研究发现,与健康对照组和其他免疫性疾病患者相比,SLE患者外周单个核细胞干扰素诱导蛋白44样蛋白(interferoninduced protein 44-like,IFI44L)基因启动子甲基化水平显著降低。IFI44L启动子甲基化在鉴别SLE方面的特异性和敏感性均优于现有的检测方法,且在区分SLE与其他自身免疫性疾病方面也具有临床应用价值。
人类基因组由可变的长末端重复序列(long terminal repeat,LTR)组成,人类内源性逆转录病毒(human endogenous retroviruses,HERVs)为LTR的组成部分,占基因组DNA的8%~9%。Okada等[80-81]研究发现SLE患者T细胞HERV-E和HERV-K基因甲基化水平降低,且其低甲基化程度与SLE活动性、抗U1-核糖核蛋白(U1-ribonucleoprotein,U1-RNP)与抗Sm抗体及补体水平呈正相关,可导致淋巴细胞减少症的发生。HERV-E和HERV-K基因低甲基化可作为区分SLE活动期和静止期的预测性指标。
3.5 DNA甲基化与镰状细胞病 镰状细胞病(sickle cell disease,SCD)为突变基因产生的脱氧镰状血红蛋白(hemoglobin sickle,HbS)在红细胞内聚合所致,可影响红细胞的活性、流变性和黏附性,促进溶血、内皮损伤、小血管闭塞和大血管血栓形成[82-83]。
有研究证明,HbS的细胞内浓度是聚合动力学的主要决定因素,采用胎儿血红蛋白(fetal hemoglobin,HbF)替代可降低HbS浓度,提示HbF可能阻断SCD的病理生理过程[84],这为药理学诱导HbF来治疗SCD提供了理论基础。
HbF中的γ-亚基由γ-珠蛋白(hemoglobin subunit gamma,HBG)基因编码,DNMT1维持DNA甲基化,参与介导HBG基因表观遗传沉默[85]。为诱导HbF表达,需抑制DNMT1对HBG基因的沉默作用。脱氧胞苷类似物地西他滨可与DNMT1共价结合,导致DNMT1降解[86]。目前口服地西他滨联合四氢尿苷(抑制地西他滨降解)治疗SCD处于临床研究Ⅰ期或Ⅱ期阶段。有Ⅰ期临床研究应用0.16 mg/kg地西他滨治疗SCD,持续8周,发现患者外周血单核细胞DNMT1下降>75%,HbF增加4%~9%,且治疗期间患者耐受良好,提示地西他滨的安全性较高[87]。该研究结果提示,地西他滨等DNMT抑制剂可作为治疗SCD新的有前景的治疗药物。
通过上面对CE、SVO、ODE型卒中与DNA甲基化关系的相关研究总结可以看出,缺血性卒中不同病因亚型具有特异性候选基因甲基化水平改变(表1)。研究候选基因DNA甲基化与非大动脉粥样硬化型卒中的关系,有助于从分子水平揭示缺血性卒中的发生、发展过程。目前该领域的研究进展已用于发现非大动脉粥样硬化型卒中相关疾病的新型生物标志物(如IFI44L甲基化用于SLE的诊断)以及发掘疾病的新型治疗靶点(如地西他滨用于SCD的治疗)。未来需要更多研究关注非大动脉粥样硬化型卒中发病的表观遗传学机制,从分子生物学角度为缺血性卒中的诊断与治疗探寻新方向。
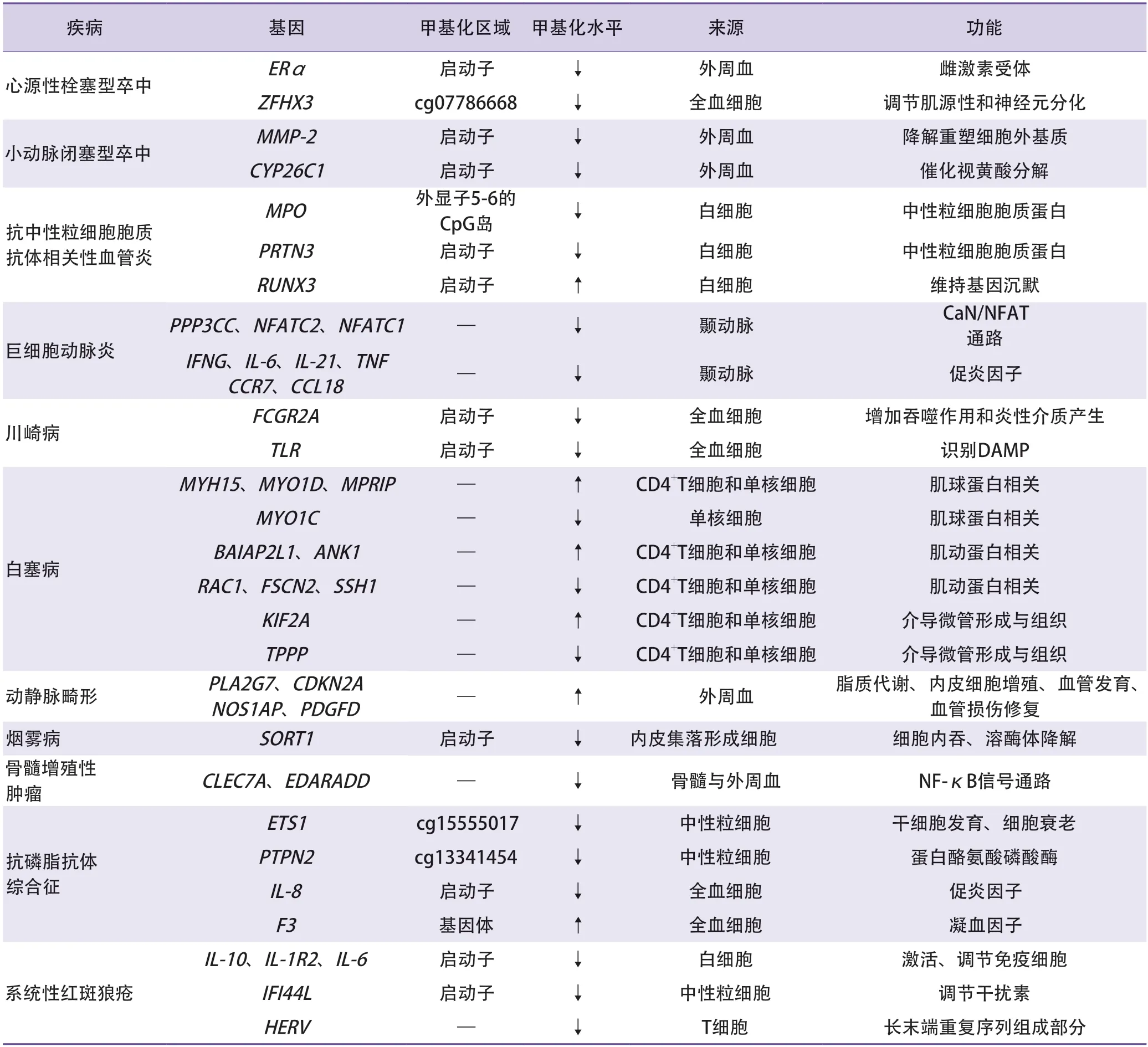
表1 非大动脉粥样硬化型卒中相关候选基因甲基化水平变化
【点睛】本文全面介绍了与心源性栓塞型、小动脉闭塞型、其他明确病因型卒中相关的DNA甲基化改变,分析其可能的机制及可能有助于临床诊断和治疗的靶点和思路。
