MEG3 基因在骨肉瘤中的研究进展
2023-05-24刘凯李宁杨浩东
刘凯 李宁 杨浩东
骨肉瘤 (osteosarcoma,OS) 是最常见的一种骨组织原发性恶性肿瘤,其病变部位主要位于长骨干骺端,临床表现为持续性疼痛并伴有夜间加重。OS 在年轻患者中发病率较高,根据美国国立卫生研究院统计,其发病率在10~19 岁时达到峰值[1]。目前在临床上治疗骨瘤的手段主要包括手术、化疗和放射治疗等[2],但因为其恶性程度较高、生长迅速且容易复发转移,因此效果大都不尽如人意。近年来随着新型辅助化疗手段的普及,患者的 5 年总存活率虽然较以往相比有显著提高,但也仅为 70% 左右,而其中发生转移和复发的患者生存率更是不到 20%[3]。
目前,OS 的发病机制尚未得到完全阐述,因此寻求新的治疗方法,提高患者的治疗率和生存率成为 OS 领域研究的热点。随着 OS 基因学说研究的深入,长链非编码 RNA (long noncoding RNA,LncRNA) 逐渐进入了人们的视线,研究显示 LncRNA 基因 MEG3 基因 (maternally expressed gene 3) 广泛存在于人体正常组织中,其在多种肿瘤组织中的低表达表明 MEG3 基因与肿瘤的发生、发展密切相关,而其在 MG-63、U-2OS 细胞系中的异常表达提示 MEG3 基因具有抑制 OS 细胞增殖、侵袭,同时促进 OS细胞凋亡的作用[4]。相关研究表明 MEG3 基因能够通过降低相关其目标靶基因的表达进一步调控 P53 蛋白从而抑制OS 的发生和发展。MEG3 基因能够通过 miR-524 和血管内皮生长因子抑制 PI3K / AKT 信号通路的传导促进 OS 细胞增殖和侵袭,调节细胞凋亡,同时推动血管的新生[5],进一步影响 OS 的发生和发展。除此之外,MEG3 基因的DNA 甲基化也与 OS 的诊疗效果密切相关,MEG3 基因的表达缺失与其 DNA 的高甲基化状态有关,可以通过降低其基因启动子的甲基化水平从而恢复 MEG3 基因的正常表达。因此,现就 MEG3 基因在 OS 中的异常表达和作用机制进行了综述,为 OS 的诊疗提供新的思路。
一、MEG3 基因的结构和功能
研究显示 LncRNA 是包含超过 200 个核苷酸但不具备编码蛋白能力的一种 RNA 分子[6],其作为调节多种生物工程中的关键因子已经成为调控 OS 发生的重要因素[7-9]。MEG3 基因作为母源表达的 LncRNA 基因长度约为 1.6 kb核苷酸,位于人类染色体 14q32.3 区域的 DLK-MEG3 印记区上[10]。基因组研究分析显示 MEG3 基因包含 10 个外显子,可通过选择性剪接产生 12 种不同的剪接体,MEG3基因通过与目标 DNA 建立 RNA-DNA 三联体从而调控其目标靶基因的表达[11-12]。
MEG3 基因作为肿瘤抑癌因子在肝癌、胃癌、肺癌、肾癌等多种肿瘤组织和细胞中表达缺失[13],研究表明,miR-29 能够通过其启动子的高甲基化状态增加 MEG3 基因的表达水平,从而抑制肝癌细胞的增殖[14];当 MEG3基因表达水平过高时还会与 miR-181s、miR-148a 以及miR-141 靶向结合,从而抑制胃癌细胞的增殖、侵袭并促进胃癌细胞凋亡[15];同时,MEG3 基因能够通过下调抗凋亡基因 Bcl-2,上调凋亡促进因子 Bax、Bad 的表达,从而抑制肺癌细胞的增殖[16-17];此外 MEG3 基因还能够通过抑制 Bcl-2 的表达,从而改变线粒体膜的通透性,激活线粒体通路,进一步诱导肾癌细胞的凋亡[18]。同时 MEG3 基因序列的多态性也与肿瘤的发生有关。MEG3 基因主要通过多种机制来调控 OS 的发展,其中包括 P53 蛋白途径、PI3K / AKT 信号通路、DNA 甲基化、Notch 和 TGF-β 信号通路等以及与 miRNAs 间的相互作用 (图1)。随着关于 MEG3 基因研究的进一步进展,其有望成为 OS 诊断、治疗和预后的潜在靶点,在 OS 的临床诊疗中发挥巨大作用,成为抑制 OS 发展的重要一环。
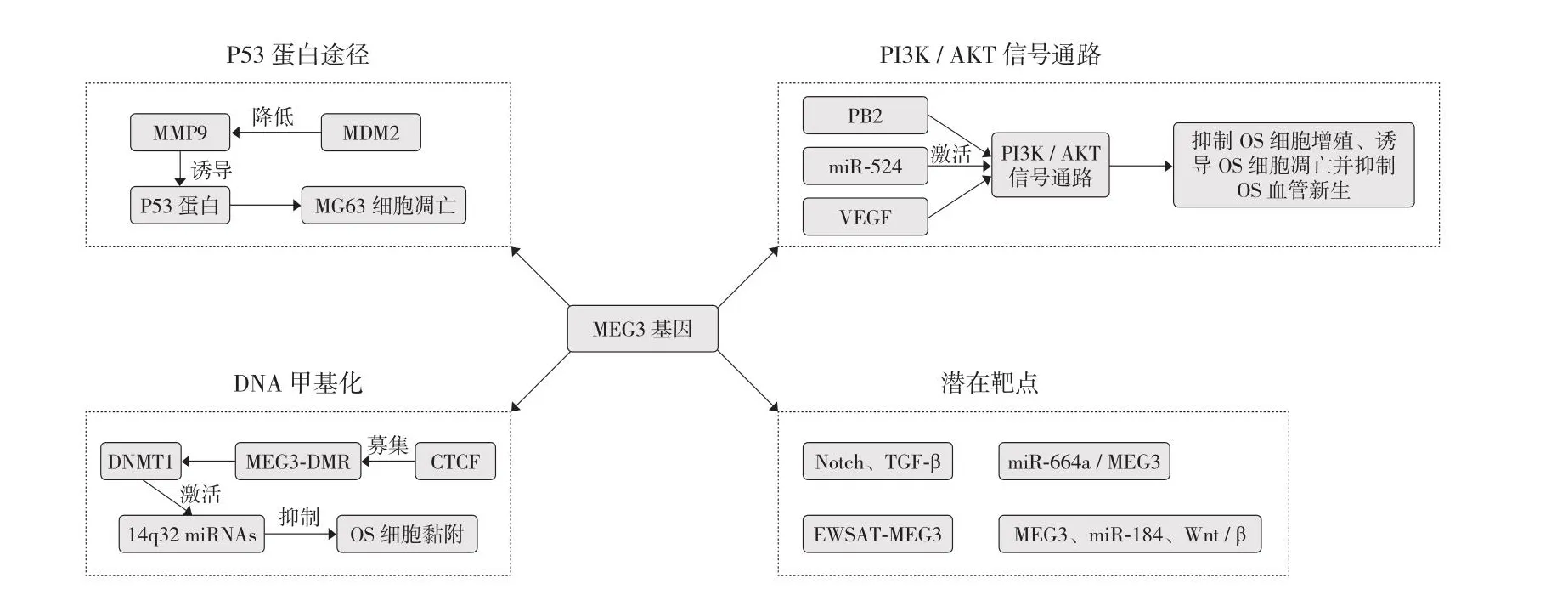
图1 MEG3 基因与 OS 作用示意图 (实线代表传导过程,虚线代表作用途径)Fig.1 Schematic diagram of the interaction between MEG3 gene and osteosarcoma (the solid line represented the conduction process, and the dotted line represented the action route)
二、MEG3 基因在 OS 中的作用机制
1.P53蛋白与 OS:P53蛋白是由 TP53 (tumorsuppressorpro-teinp53) 基因编码的一个转录因子,作为具有调节转录能力的序列特异性 DNA 结合蛋白,其主要由 6 个蛋白质结构域构成,包括两个氮末端反式激活结构域(TADs)、一个中央脱氧核糖核酸结合结构域 (DBD)、一个富含脯氨酸的结构域 (PRD)、一个四聚化结构域 (TD)和一个碳末端调节结构域 (CTD)[19]。通过与位于靶基因启动子或增强子的 P53 应答元件结合可以激活多个基因的表达,因此被称为“基因组的守护者”[20-21]。在正常情况下,P53 蛋白通过 MDM2 (murine / humandoubleminute 2)介导的 P53 泛素连接酶降解从而保持在极低的水平[22],但同时可以通过脱氧核糖核酸损伤、代谢缺损、缺氧等多种细胞应激源激活[23]。研究显示,MEG3 基因过表达时能够提高 P53 蛋白的稳定性并通过多种途径调控 P53 蛋白的表达,进一步调控肿瘤的生长发育。MEG3 基因在 OS 中的表达明显减少,这与其生存率较低且容易发生远端转移密切相关[24]。MEG3 基因的上调能够通过降低 MDM2 的表达及其目标靶基因 MMP9 的表达诱导 P53 蛋白的表达从而导致 MG63 人 OS 细胞凋亡,抑制人的 OS 细胞繁殖、侵袭,进一步抑制 OS 的发展[25]。
综上所述,P53 作为肿瘤抑制因子能够调节众多靶基因的表达,而 MDM2 则是 P53 表达的主要抑制因子。MEG3 基因一方面通过 MDM2 与 P53 的 N 端结构域结合掩盖了 P53 对转录机制的访问并通过 MDM2 使 P53 泛素化,从而针对 P53 进行蛋白酶体降解。提示 MEG3 基因可以通过 MDM2 激活 P53 进一步抑制 OS 细胞的增殖、侵袭,但其具体机制仍须进一步研究。
2.PI3K / AKT 信号通路与 OS:磷脂酰肌醇 3-激酶(phosphatidylinositol 3 kinase,PI3K) 和蛋白激酶 B (protein kinase B,AKT) 作为受生长因子、细胞因子等多种因素参与调控的信号通路,广泛存在于各种细胞中,并参与细胞生长繁殖、转录和存活等多种细胞活动[26-27]。其中PI3K 作为同时具有磷脂激酶和蛋白激酶两种活性的胞内磷脂酰肌醇激酶,根据其结构特点可分为 Ⅰ 型 PI3K 亚型(PI3Kα,β,γ)、Ⅱ 型 PI3K 亚型 (PI3KC2α,C2β,C2γ)以及 Ⅲ 型 PI3K[28]。其中Ⅰ 型 PI3K 是由催化亚单位(PIK3CA、PIK3CB、PIK3CD 或 PIK3CG) 和一个调节亚单位组成的异二聚体激酶,由于容易被细胞表面受体活化,因此与肿瘤的发生、发展紧密相连。Ⅰ 型 PI3K 通过磷酸化磷脂酰肌醇-4,5-二磷酸 (PI-4,5-P) 肌醇环的3’-羟基,转导由受体酪氨酸激酶 (RTKs) 和 G 蛋白偶联受体 (GPCRs) 产生的上游信号进而生成磷脂酰肌醇-3,4,5-三磷酸盐 (PIP3),作为关键的脂质第二信使,PIP3 将含有 pleckstrin homology (PH) 结构域的胞质蛋白集中到质膜上,以促进其活化或与其它效应蛋白共定位[29-30]。研究表明,Myc 是上调细胞周期蛋白依赖激酶 (CDK) 4 的癌蛋白,PI3K 能够通过增加 c-Myc 的表达水平并促进 OS 细胞的增殖和侵袭[31]。而 AKT 作为 PI3K / AKT 信号通路的关键环节包含三个结构域,分别为 pleckstrin 同源性结构域、中间激酶结构域以及调节性羧基末端结构域,根据氨基酸残基的不同可将 AKTs 分为三种亚型,分别为 AKT1型、AKT2 型和 AKT3 型。AKT 主要通过两个关键的磷酸化过程激活,第一步由磷酸化的苏氨酸 308 (AKT1) 在激酶结构域 phosphoinositide-dependent 蛋白激酶 (PDK1) 启动活化过程,随后通过 mTOR 复合物 2 (mTORC2) 在羧基末端调节结构域的丝氨酸 473 (AKT1) 磷酸化,该过程被PI3K 依赖机制激活并完全激活 AKT[32]。同时,含有 PIP3和 PIP2 的内膜也被证明直接有助于 AKT 的激活[33-34]。AKT 在抑制细胞凋亡方面起着至关重要的作用,能够通过降低 Bad 和 Bax 的表达水平,提高 Bcl-2 和 Bcl-xl 的表达水平抑制 OS 细胞的增殖[35]。
MEG3 基因依靠 PI3K / AKT 信号通路在 OS 细胞中的活跃表达,减少 OS 细胞的增殖、侵袭并诱导其凋亡[36]。研究显示,磷酸酶和张力蛋白同系物 (PTEN) 作为一种具有生长和调节功能的负调节因子可以阻断 PI3K / AKT 信号通路的传导[37],而 MG3 基因可以通过激活 miR-524 进一步抑制 PTEN 从而启动 PI3K / AKT 信号通路的传导,进一步促进 OS 细胞的增殖和侵袭[38-39];Wu 等[40]通过用不同浓度的原花青素 (PB2) 处理 OS 细胞株及 OB 细胞,结果显示,PB2 可以通过切断 PI3K / AKT 信号通路从而抑制 OS细胞的增殖并诱导 OS 细胞凋亡。同时 PI3K / AKT 信号通路在推动血管新生方面也起着不可估量的作用,血管内皮生长因子 (vascular endothelial growth factor,VEGF) 能够促进血管生成、血管重塑并且增加血管通透性。研究表明,MEG3 基因表达水平降低会抑制 VEGF 的表达,而 VEGF沉默又可以通过阻断 VEGF / PI3K / AKT 信号通路抑制 OS细胞增殖、减少 OS 血管的生成并促进 OS 细胞凋亡[41-42]。
由此可得出 MEG3 基因可以通过控制 PTEN、PB2、VGEF 的表达水平抑制 PI3K / AKT 信号通路的传导从而抑制 OS 细胞的增殖、诱导 OS 细胞凋亡并减少 OS 血管的新生,同时,MEG3 基因的异常表达还可以通过PI3K / AKT 信号通路降低 Bad、Bax 的表达水平并上调Bcl-2、Bcl-xl 的表达水平,进一步抑制 OS 细胞的迁移和侵袭,在 OS 的发生、发展过程中发挥重要作用,为临床上治疗 OS 提供新的思路。
3.DNA 甲基化与 OS:研究发现,MicroRNA (miRNA)作为由内源基因编码的单链 RNA 分子通过与靶基因 3’ 非翻译区碱基配对抑制翻译并切割 mRNA,能够参与转录后的基因表达调控,具有调节肿瘤细胞增殖、分化、凋亡的能力,而由人类染色体 14q32 区域编码的的 miRNAs正是 OS 患者的良好预后因素[31,43-45]。DNA 甲基化作为调控 MEG3 基因表达的表观遗传事件之一,主要通过脱氧核糖核酸甲基转移酶 (DNMTs) 在胞嘧啶鸟嘌呤二核苷酸 (CpG) 的环境中表达发挥作用并参与 14q32 miRNAs 的表达调控[46]。Katherine 等[47]通过对 19 个 OS 细胞系用miRNA (Agilent 阵列) 和甲基化数据 (Illmina 27K 阵列) 进行检测,发现多个 CpG 位点在侵袭型、转移性、致癌性较高的细胞系中甲基化程度更高。同时 Oshima 等[48]研究表明,DNA 去甲基化通过促进 ccctc 结合因子 (CTCF) 募集到 MEG3 基因差异甲基化区 (MEG3-DMR),而 MEG3-DMR 作为 14q32 miRNAs 表达的顺式调控元件,能够通过DNMT1 激活 MEG3-DMR 的去甲基化和 14q32 miRNAs 的表达,从而抑制 OS 细胞的黏附、侵袭和转移特性。
综上所述,OS 中 MEG3 基因表达水平降低,可能与其基因启动子的甲基化水平过低有关,可以通过其启动子的高甲基化状态增加 MEG3 基因的表达水平,进一步激活14q32 miRNAs 的表达从而抑制 OS 细胞的增殖、侵袭并诱导 OS 细胞凋亡,表现出抑制 OS 发展的能力。
三、MEG3 基因在 OS 临床治疗中的价值
研究发现,MEG3 基因作为 OS 诊断和预后的生物标志物有望成为一种潜在的新型治疗靶点来缓解 OS 患者的痛苦。Li 等[49]通过测量 pcDNA-MEG3 联合传导后的小鼠肿瘤体积并与空白组进行对照,同时对 Wnt / β-连环蛋白进行蛋白质印迹法检测,发现 MEG3 能够抑制miR-184 的表达,同时用 pcDNA-MEG3 联合传导的实验组中小鼠肿瘤体积明显减小且存活率增加。提示 MEG3 可以通过靶向 miR-184 抑制并下调 Wnt / β-连环蛋白通路从而抑制 OS 的发生、进展和转移。Zhang 等[50]通过检测与细胞生长转移相关的因子,发现当 MEG3 高表达时能够抑制 Notch1、Hes1、TGF-β 和 N-cadhern 的表达水平,同时提高 E-cadhern 的表达水平。提示 MEG3 基因作为 OS 临床治疗的新靶点能够通过抑制 Notch 和 TGF-β 信号通路抑制 OS 细胞的增殖和转移。Sahin 等[51]通过 HiPerFect 转染试剂将 miR-664a 抑制剂分别转染于 OS 细胞株 (U2-OS)和 OB 细胞株 (hFOB 1.19) 中,发现 MEG3 与 miR-664a的表达水平呈反比关系,同时抑制 miR-664a 能显著增加 MEG3 的表达并且通过 MEG3 和 miR-664a 之间的相互作用影响 OS 细胞的转移,提示 miR-664a 作为一种onco-microRNA 能够抑制 MEG3 信号在 OS 中的传导,并且 miR-664a / MEG3 可能作为一种新型标志物和潜在的治疗靶点在 OS 的临床诊疗中发挥作用。同时 Sun 等[52]发现EWSAT1 也能通过抑制 MEG3 基因的表达进一步促进 OS细胞的增殖和转移,提示 EWSAT1-MEG3 也可能是 OS 临床治疗的潜在靶点。目前针对 MEG3 基因在 OS 的临床治疗中具有治疗意义的新型潜在靶点缺乏相关的临床试验,其准确性和治疗效果有待进一步研究。
四、结语和展望
OS 是一种恶性程度较高、生长迅速且容易复发转移的原发性骨骼恶性肿瘤,其发病原因多与 OS 细胞的异常增生及基因突变有关。目前临床上主要通过手术、热疗、化疗和放疗等手段来治疗 OS,但临床疗效较差且容易复发。MEG3 在 OS 组织及细胞系中表达缺失与其 DNA 的高甲基化状态密切相关,当 MEG3 基因表达缺失时,可能会促进 OS 细胞的增值、侵袭并抑制 OS 细胞凋亡,从而参与 OS 的发生与发展;而当 MEG3 基因表达过度时则可能会通过其相关表观遗传学调控 P53 蛋白途径、PI3K / AKT信号通路、Notch 和 TGF-β 信号通路以及与 miRNAs 的相互作用抑制 OS 的发展。目前研究显示 MEG3 基因有望成为 OS 诊断、治疗和预后的新型潜在靶点并且已经成为 OS临床研究领域中的热点。虽然目前的研究成果已经证明了MEG3 基因在 OS 治疗中的重要性,但其在 OS 中的作用机制和潜在的调控路径还需要进一步的挖掘和探索,相信随着研究的不断深入,以上问题将会得到解决,从而为在临床上应用 MEG3 基因治疗 OS 提供更加可靠的理论依据。
