体外受精胚胎移植婴儿先天性聋临床和基因检测分析
2023-05-22胡民强谭东辉杨曙朱倩晨赖若沙谢华平邓健航邓忠谢鼎华伍伟景
胡民强 谭东辉 杨曙 朱倩晨 赖若沙 谢华平 邓健航 邓忠 谢鼎华 伍伟景*
1 中南大学湘雅二医院耳鼻咽喉头颈外科(长沙 410011)
2 中南大学耳科研究所(长沙 410011)
3 湘南学院附属医院(郴州 423000)
4 湖南师范大学生命科学学院(长沙 410081)
5 中南大学湘雅医学院附属株洲医院耳鼻咽喉头颈外科(株洲 412000)
越来越多的夫妻通过辅助生殖技术来帮助怀孕,其中体外受精胚胎移植(in vitro fertilization and embryo transfer,IVF-ET)最为常见,通常被称为“试管婴儿”技术[1]。临床上试管婴儿技术不仅用于解决生育障碍的困境,对于某些单基因遗传病可通过体外阻断,达到优生优育的目的。试管婴儿并非自然生殖,研究发现通过辅助生育技术育出的婴儿出生缺陷的几率比自然受孕的婴儿要高[2],这些缺陷的出现是受到辅助生育技术处理流程的影响,还是由遗传因素所致,目前尚无明确结论[3]。
临床上时常遇到,采取IVF-ET 技术妊娠的夫妻,即使双方没有耳聋病史和家族史,也会生育出先天性聋的婴儿。本文对3 个IVF-ET 先天性聋家庭进行了详尽的临床资料收集、遗传特征分析及遗传学检测,对包括4 名先天性感音神经性聋(sensorineural hearing loss,SNHL)患儿在内的5 名婴儿及其父母进行了全外显子组基因测序(whole exome sequencing,WES)和Sanger测序分析,以期为明确这些婴儿出现先天性SNHL 的原因,避免IVFET生育听力缺陷婴儿提供指导。
1 资料与方法
1.1 临床资料
本研究收集2020年1月至5月在中南大学湘雅二医院就诊的3 个IVF-ET 先天性耳聋家庭,均为父母亲本供精供卵。3 个家庭共生育5 名婴儿,家庭1 和家庭2 为异卵孪生,足月剖宫产,家庭3 为单胎,足月顺产。其中家庭1男婴和女婴,家庭2女婴,家庭3 男婴出生时新生儿听力筛查均未通过,家庭2男婴听力筛查通过。
对3 个家庭成员的家族史、遗传史、临床表现等资料进行收集,排除相关病毒感染、缺氧及病理性黄疸等致聋病因,对所有婴儿及其父母共11 名成员分别进行相关耳科和全身检查,全面听力学评估,签署知情同意书后,采取外周静脉血进行遗传基因检测。婴幼儿听力评估采取听性脑干反应、多频稳态反应、耳声发射和声导抗测试为主,结合声场测听进行主观听力评估;成人听力评估采取纯音测听、声导抗和耳声发射测试。确诊为双耳极重度SNHL 的4 名婴儿均进行耳部高分辨率CT 和磁共振成像检查,并接受人工耳蜗植入。该研究得到中南大学湘雅二医院伦理委员会批准。
1.2 研究方法
1.2.1 全外显子组测序
采用基因组DNA 提取试剂盒提取基因组DNA,经Qubit3.0 荧光定量仪鉴定,所提取的DNA浓度和纯度合格,可用于后续实验。
应用基于目标序列捕获的二代测序技术进行WES 测序鉴定。实验流程包括:文库构建,分选纯化DNA及片段化,末端修复,3’末端加A,连接测序接头,文库纯化和片段大小筛选,PCR扩增文库,全外显子芯片杂交,杂交文库清洗及纯化,PCR 扩增外显子DNA文库,文库质量检测,上机测序。
1.2.2 生物信息分析
对测序数据进行汇总,获取每个样本原始序列数量和测序读长,以“Q20(%)”和“Q30(%)”分别表示原始数据中测序质量值Q 不低于20和30的序列碱基比例。通过对三组家庭进行WES,分析发现数据量在13GB~18GB 之间,平均测序深度在155X,目标测序区域10X 覆盖碱基覆盖比例约99%,Q20 的比例均在97%以上,Q30 的比例在92%以上。
碱基质量评估,比对,使用BWA 工具将测序序列比对到hg19。比对完成使用Picard 工具进行Q20,Q30 平均测序深度等数据统计。使用GATK Haplotype Caller 方法检测样本的SNV/InDel。通过annovar 工具对突变进行基因功能等注释。运用SNPscan 分型技术检测96个高频位点,验证全外显子组测序的准确性。
1.2.3 致病基因筛选
针对全部的SNV/InDel 位点,保留符合这些条件的位点用于进一步分析:①频率性数据库选择低频突变:1000Genomes的频率低于0.01;②属于外显子突变或剪切位点突变的所有不是同义突变的位点;③突变在保守性数据库(Sift,POLYPhen V2,Mutation Taster,Cadd,Dann,dbscSNV)中预测为有害;④基于基因功能以及基因与疾病筛选和耳聋相关的基因。
1.2.4 Sanger测序验证
在WES 的基础上对10 个样本的7 个位点进行了测序验证,总共设计了7 对引物。①DNA 样本取1l 1%agarose 电泳对其样本进行质量及浓度估计,再根据浓度将样本稀释到工作浓度5-10ng/l,没有DNA 条带的样本保持原浓度。②设计PCR 反应,包括PCR 引物和设计PCR 条件。③利用SAP 和Exo I 进行PCR 纯化。④测序产物上ABI3730XL 测序仪,测序文件用Polyphred 软件分析,最后人工校对,整理出结果。
2 结果
2.1 临床结果
3 个家庭父母听力均正常,家族中无耳聋患者。家庭1母亲受孕时31岁,有宫外孕史,患“甲状腺功能减退”8年。家庭2 母亲受孕时29 岁,患“多囊卵巢综合征”,月经紊乱。家庭3 母亲受孕时32岁,患“输卵管不通,多囊卵巢综合征”,月经紊乱,有不明原因流产史。
听力评估结果表明,家庭1 双胞胎男婴和女婴均为双耳极重度SNHL;家庭2 双胞胎中男婴听力正常,女婴为双耳极重度SNHL;家庭3 男婴为双耳极重度SNHL。
影像评估显示家庭1 两名耳聋婴儿内耳发育无畸形,家庭2 耳聋女婴为Mondini 畸形并前庭水管扩大(图1)。家庭3 耳聋男婴双侧内听道狭窄(图2)。

图1 家庭2聋儿耳部CT显示Mondini畸形并前庭水管扩大。Fig.1 CT scan of deaf infant in Family 2 showing Mondini malformation and enlarged vestibular conduct.
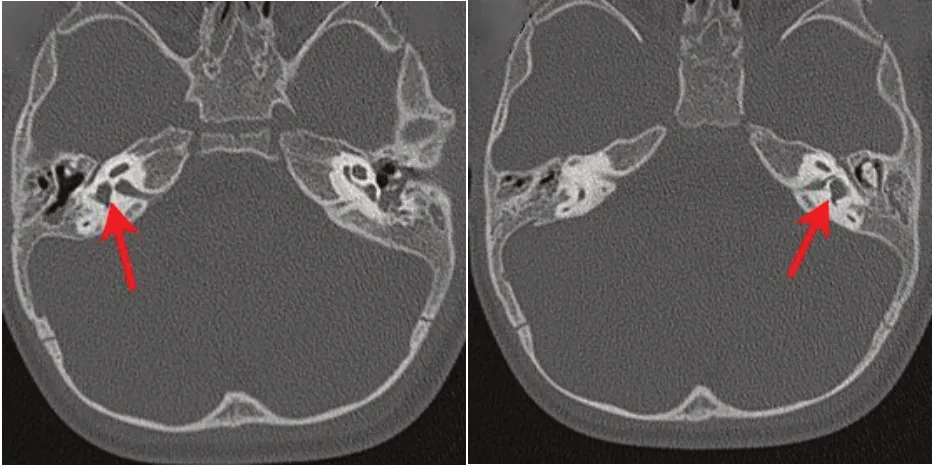
图2 家庭3聋儿耳部CT显示内听道狭窄。Fig.2 CT scan of deaf infant in Family 3 showing stenosis of internal auditory canal.
2.2 基因检测结果
运用WES 对约20000 个基因测序后发现突变候选基因3193个,分析筛选出4个常见与耳聋相关基因,其他为目前认为与耳聋无相关性的隐性基因和新生突变基因。
2.2.1 三组家庭耳聋相关基因突变
三组家庭11 名成员WES 基因检测共发现4 个基因7 个突变位点,包括MYO3A、MYO15A、SLC26A4和TPO基因(表1)。
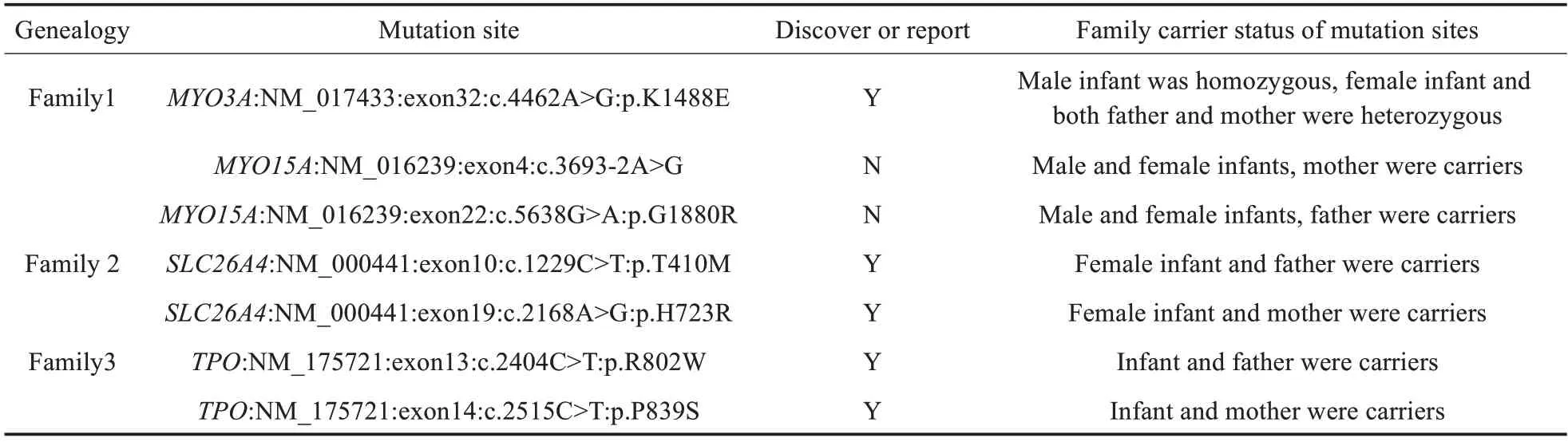
表1 基因检测结果(人),n=11Table 1 Genetic test results(human),n=11
2.2.2 三组家庭父母及患儿的耳聋基因突变位点
家庭1患儿父母均为MYO3A杂合突变,其突变位点相同,为MYO3A:NM_017433:exon32:c.4462A>G:p.K1488E(图3A,B)。其父母同时还携带突变位点不同的MYO15A杂合突变,父亲突变位点为:MYO15A:NM_016239:exon22:c.5638G>A:p.G1880R(图4A);母亲突变位点为:MYO15A:NM_016239:exon4:c.3693-2A>G(图4B),该两处突变位点位于剪切区,不参与蛋白质的合成,过去未见报道。
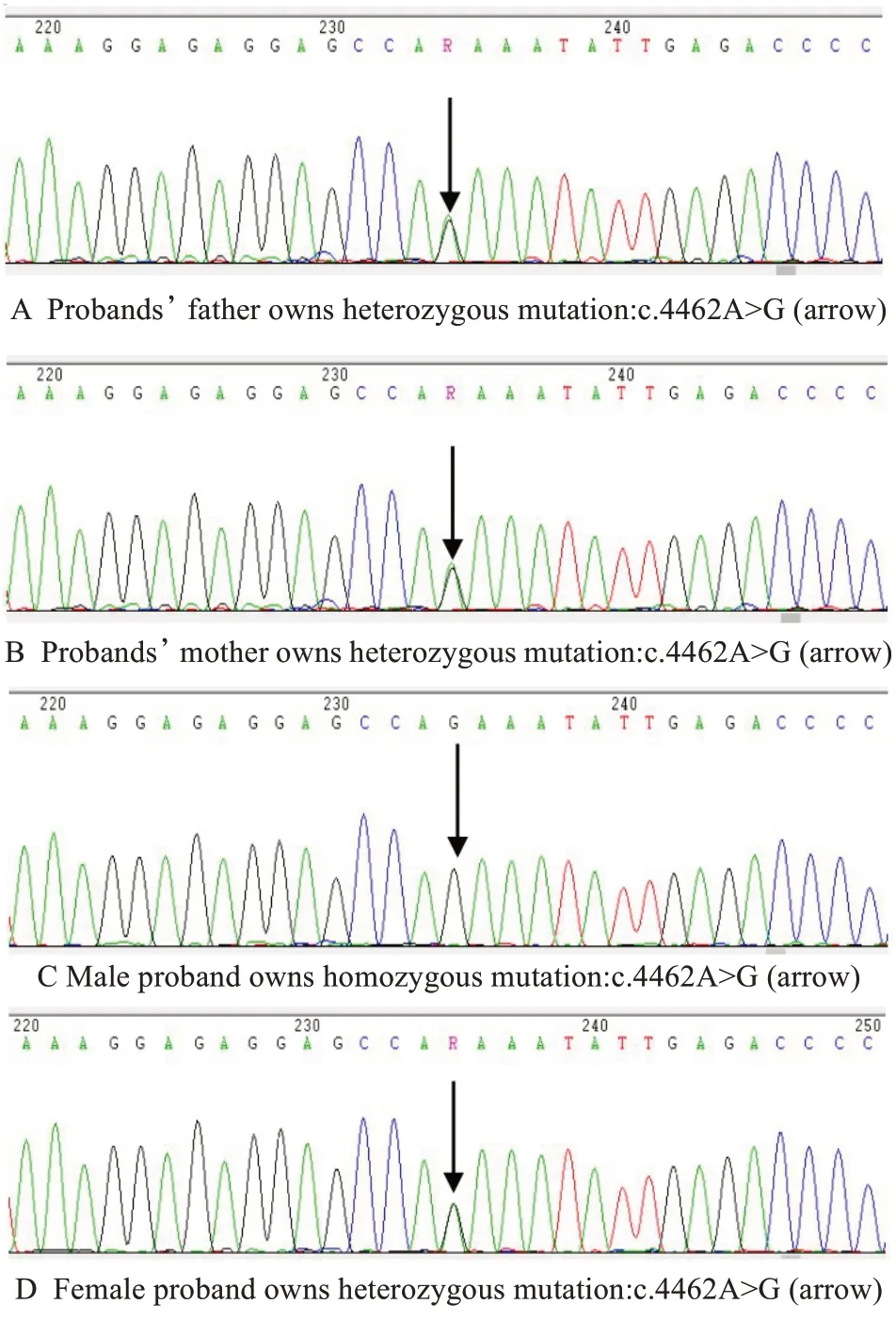
图3 家庭1 MYO3A 基因测序图。A:先证者父亲拥有杂合突变:c.4462A>G;B:先证者母亲拥有杂合突变:c.4462A>G;C:男先证者拥有纯合突变:c.4462A>G;D:女先证者拥有杂合突变:c.4462A>G。Fig.3 MYO3A gene sequencing of Family 1.A:Proband's father owns heterozygous mutation:c.4462A>G ; B: Proband's mother owns heterozygous mutation:c.4462A>G; C: Male proband owns homozygous mutation:c.4462A>G; D: Female proband owns heterozygous mutation:c.4462A>G.
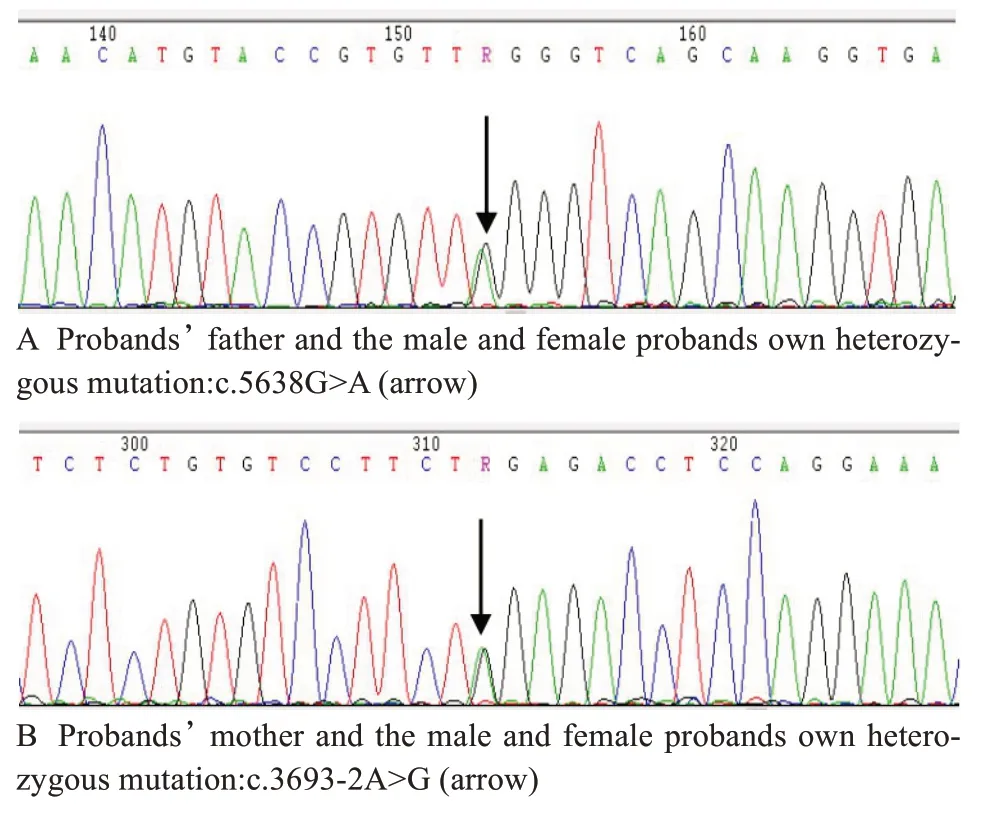
图4 家庭1 MYO15A 基因测序图。A:先证者父亲和男女患儿拥有杂合突变:c.5638G>A;B:先证者母亲和男女患儿拥有杂合突变c.3693-2A>G。Fig.4 Gene sequencing of Family 1 MYO15A.A: Proband's father the male and female probands own heterozygous mutation:c.5638G>A; B: Proband's mother and the male and female probands own heterozygous mutation:c.3693-2A>G.
家庭1 男患儿为MYO3A基因纯合突变,与父母共有突变位点MYO3A: NM_017433: exon32:c.4462A>G:p.K1488E(图3C),同时还携带其父母的MYO15A突变位点,表现为复合杂合突变,与母亲共有突变位点MYO15A:NM_016239:exon4:c.3693-2A>G,与父亲共有突变位点MYO15A:NM_016239:exon22:c.5638G>A:p.G1880R。女患儿为MYO3A基因杂合突变,突变位点与其父母相同(图3D),同时也携带来自其父母的MYO15A基因突变位点,表现为复合杂合突变,与其父亲共有突变位点MYO15A:NM_016239:exon22:c.5638G>A:p.G1880R,与母亲共有突变位点MYO15A:NM_016239:exon4:c.3693-2A>G。
家庭2 中父母均为SLC26A4杂合突变,但双方突变位点并不相同。父亲突变位点为:SLC26A4:NM_000441:exon10:c.1229C>T:p.T410M(图5A);母亲突变位点为:SLC26A4: NM_000441: exon19:c.2168A>G:p.H723R(图5B)。家庭2 中女患儿携带来自父母的SLC26A4基因突变位点,表现为复合杂合突变;与其父亲共有的突变位点为:SLC26A4:NM_000441:exon10:c.1229C>T:p.T410M;与其母亲共有的突变位点为:SLC26A4:NM_000441: exon19:c.2168A>G:p.H723R。家庭2 中另一男婴经筛选为野生型。
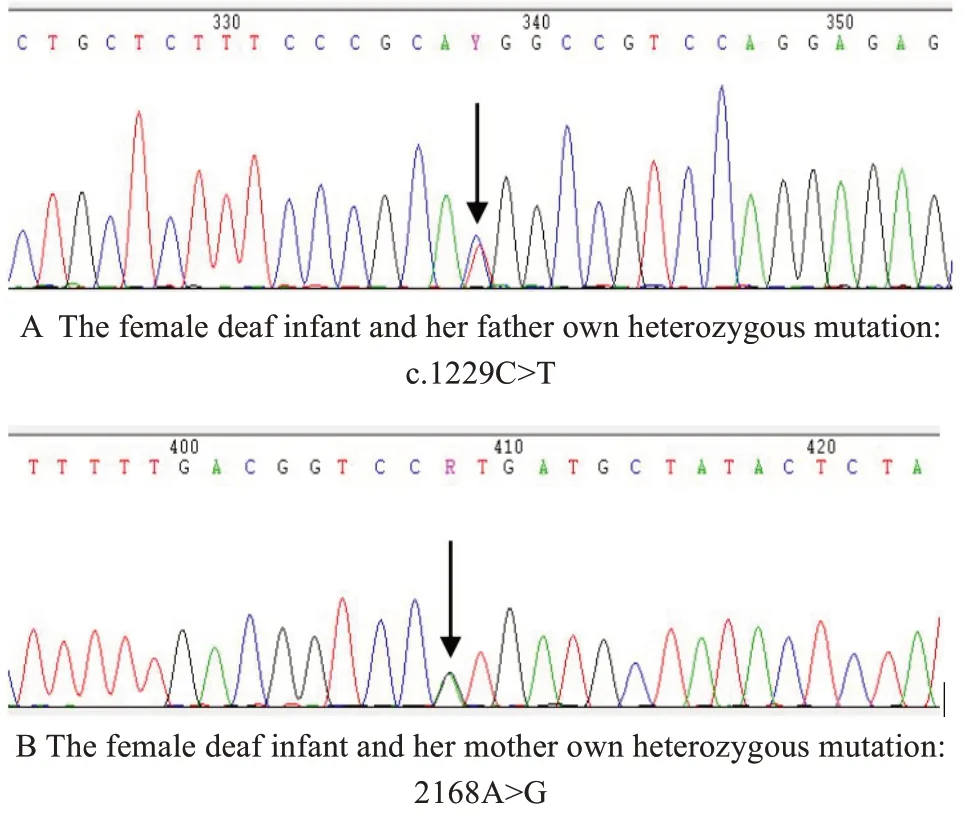
图5 家庭2 SLC26A4 基因测序图。A: 患儿和父亲共有杂合突变:c.1229C>T;B:患儿和母亲共有杂合突变:2168A>G。Fig.5 Family 2 SLC26A4 gene sequencing.A: The female deaf infant and her father own heterozygous mutation:c.1229C>T; B: The female deaf infant and her mother own heterozygous mutation:2168A>G.
家庭3 中父母均为TPO杂合突变,双方突变位点不同。父亲突变位点为:TPO:NM_175721:exon13:c.2404C>T:p.R802W(图6A);母亲突变位点为:TPO:NM_175721:exon14:c.2515C>T:p.P839S(图6B)。家庭3 患儿携带来自其父母的TPO基因突变,表现为复合杂合突变。与父亲共有的突变位点为TPO:NM_175721:exon13:c.2404C>T:p.R802W;与母亲共有的突变位点为TPO:NM_175721:exon14:c.2515C>T:p.P839S。
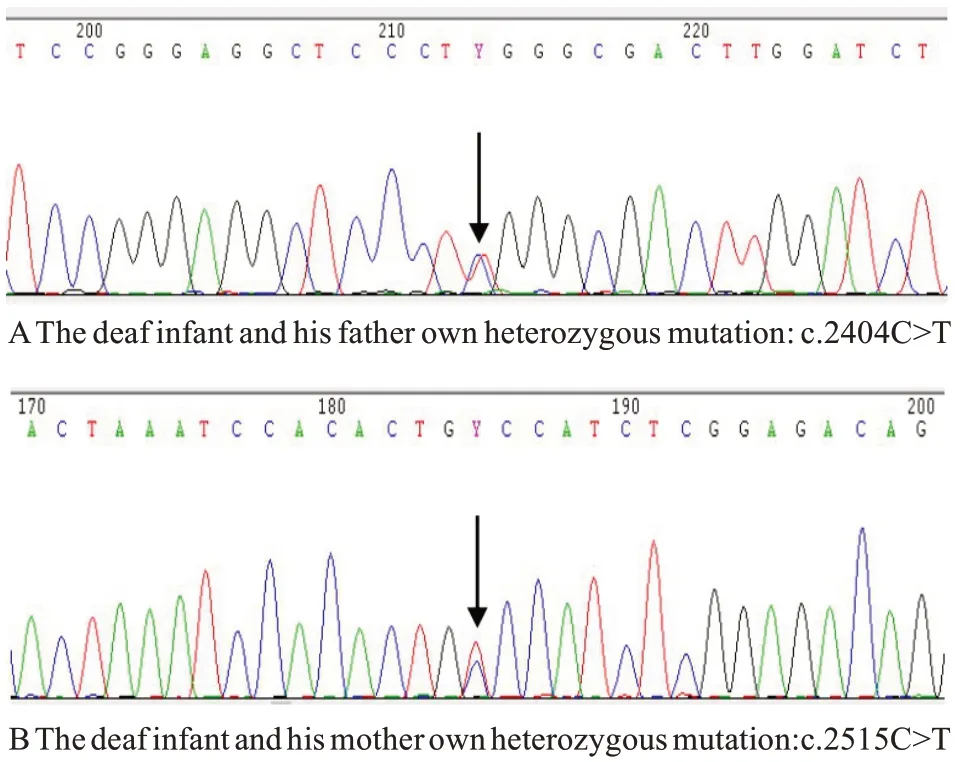
图6 家庭3 TPO 基因测序图。A:患儿和父亲共有杂合突变:c.2404C>T;B:患儿和母亲共有杂合突变:c.2515C>T。Fig.6 TPO gene sequencing of family 3.A: The deaf infant and his father own heterozygous mutation: c.2404C>T; B:The deaf infant and his mother own heterozygous mutation:c.2515C>T.
本组病例WES基因测序表明,家庭1男患儿携带耳聋相关突变基因MYO3A,为纯合突变,同时携带MYO15A突变基因,为复合杂合突变。女患儿携带MYO15A突变基因,为复合杂合突变。家庭2 中女患儿携带SLC26A4基因突变位点,为复合杂合突变。家庭3 中男患儿携带TPO突变基因,为复合杂合突变。以上突变均由其父母提供,所有先证者其耳聋的产生符合遗传规律。
3 讨论
临床上通过辅助生殖技术生育出先天性缺陷婴儿并非少见,逐渐引起人们关注。不孕不育会增加试管婴儿出生缺陷的风险,国外研究报道IVFET 或胞浆内单精子注射(intracytoplasmic sperm injection,ICSI)的出生缺陷率分别为8.6% 和9.0%[3],但另一项研究指出在考虑包括母亲年龄在内的父母因素的多变量分析后,IVF-ET 和出生缺陷之间并无明显关联[4]。辅助生殖技术伴随着出生缺陷风险增加,可能涉及的因素包括潜在的低生育能力、诱导排卵药物、显微操作技术等[5]。
目前国内外关于IVF-ET与出生缺陷的研究较多,但对其与听力缺陷的关系报道较少。Ludwig等[6]通过前瞻性、对照、盲法随访研究发现运用ICSI与自然受孕对照组的听力和视力发育无差异。而Liang 等[7]报道,与自然受孕相比,IVF-ET 或ICSI 技术单胎妊娠眼、耳、脸和颈部畸形的风险增加20%。Kim 等[8]报道IVF-ET 技术导致听力筛查失败可能有以下几个原因,如IVF-ET 技术程序本身的直接影响,或者与IVF-ET技术程序相关的处理(如辅助孵化、囊胚培养)以及与不孕症本身有关。研究者发现运用IVF-ET或ICSI技术可能引起基因突变导致异常甲基化,进而引起表型的改变,并报道RIPOR2突变引起甲基化异常可能与听力缺陷有关,因为它编码毛细胞纤毛的质膜相关蛋白[9]。齐月娥等[10]报道,试管婴儿与正常妊娠出生的新生儿听力检查结果无明显统计学差异,孟晔等[11]通过基因测序发现运用IVF-ET或ICSI技术子代遗传性耳聋基因突变发生率与自然妊娠组子代相似。我们在近年的临床实践中,发现IVF-ET 生育先天性聋儿的实例并非罕见,而且在这些家系中,父母及家族成员均无耳聋,亦未调查出遗传家族史。
基于在遗传性聋中,非综合征性隐性遗传所占比例最高,在考虑辅助生殖技术伴发先天性聋的病因时,遗传因素必须引起高度重视。虽然这些家庭可能没有耳聋家族史,父母听力表现正常,但其携带隐性遗传耳聋基因的可能性值得注意。在本组IVF-ET 先天性聋病例的遗传学探查与分析中,根据文献报道及在线人类孟德尔遗传数据库(Online Mendelian Inheritance in Man,OMIM)、人类遗传学数据库(Human Gene Mutation Database HGMD)等数据库筛选出4 种耳聋相关致病基因MYO15A、MYO3A、SLC26A4、TPO,这4 种基因突变与三组家庭出现耳聋患儿的相关性极大。值得注意的是,家庭1 同时存在MYO3A和MYO15A两种耳聋相关基因突变,根据ACMG 指南MYO3A:NM_017433:exon32:c.4462A>G:p.K1488E 变异的致病性分级为良性,提示该突变位点可能不是致聋原因,但Wu等[12]曾报道该基因纯合突变的感音神经性聋。另外,MYO15A的2 个新突变位点的致病性有待进一步验证,可对新发现的突变位点进行克隆,进一步构建载体,再通过显微注射到胚胎,或者通过CRISPR/Cas9 基因敲出技术摸拟突变位点,观察表型的改变。TPO基因突变被认为是自身免疫性甲状腺疾病的常见原因,Kara 等[13]报道TPO突变可能与甲状腺功能减退症伴先天性聋有关。本组家庭3 患儿未检测出甲状腺功能异常,需追踪观察,其TPO基因突变与耳聋的关系待进一步验证。
本组病例中,除了上述4 种耳聋相关基因外,还发现三个家庭均携带有其他隐性遗传突变基因,如家庭1 中还有CYP1A1纯合突变,DMXL1、HMCN2、MUC16复合杂合突变,家庭2 有PDE4DIP纯合突变,HERC2、HMCN2、MIA3、TGFBRAP1、OBSCN、TTC37复合杂合突变,家庭3 有AHNAK复合杂合突变。通过查阅相关文献和OMIM、HGMD数据库,这些基因突变与耳聋关系不大,可能参与其他表型[14]。
总之,3 组家庭父母携带有MYO15A、MYO3A、SLC26A4、TPO与耳聋相关的基因突变,患病婴儿的临床表现符合遗传规律。因此,临床上开展辅助生殖技术时,与自然受孕一样,应高度重视对候选夫妻进行广泛耳聋基因的筛查,且不仅限于目前常见的4 种基因,做好产前诊断,以减少听力缺陷婴儿的出生,达到优生优育的目的。
