黄背草与2种菅属植物叶绿体基因组特征比较及系统发育分析
2023-05-20赵鹏宇侯智扬刘博宽谢睿琪曹泽林王开开
赵鹏宇,赵 威,侯智扬,李 潇,刘博宽,杜 瑶,谢睿琪,曹泽林,王开开
• 药材与资源 •
黄背草与2种菅属植物叶绿体基因组特征比较及系统发育分析
赵鹏宇1, 2,赵 威1*,侯智扬1,李 潇1,刘博宽1,杜 瑶1,谢睿琪3,曹泽林1,王开开1
1. 河南科技大学农学院,河南 洛阳 471000 2. 中国科学院分子植物科学卓越创新中心 植物分子遗传国家重点实验室,上海 200032 3. 河南科技大学基础医学与法医学院,河南 洛阳 471000
以黄背草为试验材料,比较分析其与阿拉伯黄背草、中华菅2种同属植物的叶绿体基因组特征及其与近缘物种的系统发育关系。采用Illumina HiSeq高通量测序平台首次对黄背草叶绿体基因组进行测序,使用SPAdes和CpGAVAS2分别对其进行组装和注释,并用CodonW、DnaSP和MISA等对其与2种同属植物进行一系列比较基因组分析,利用最大似然法(maximum likelihood,ML)构建系统进化树。3个叶绿体基因组全长为138 735~138 961 bp,具有典型的四分体结构,共注释出129个基因;黄背草与其同属的2个物种相比,反向重复区(inverted repeats,IR)收缩了2132 bp,大单拷贝区(large single copy region,LSC)扩张了约4000 bp,而小单拷贝区(small single copy region,SSC)变化不大。密码子偏好性分析显示,3个叶绿体基因组相对丰度最大和最小的密码子都相同,同义密码子相对使用丰度略有不同,但差异不大。核酸多态性分析显示,3个叶绿体基因组间区序列的核酸多态性值(Pi)普遍高于共有基因序列,且IR区域相比于LSC、SSC区域更为保守。3个叶绿体基因组中分别检测到38、36、37个简单重复序列(simple sequence repeat,SSR)和2种长重复序列(long sequence repeat)。系统发育分析表明,黄背草与阿拉伯黄背草亲缘关系最近。对黄背草与2种菅属植物叶绿体基因组特征及其系统发育进行了比较分析,为上述3种植物的物种鉴定及菅属遗传多样性和系统进化研究奠定了基础。
黄背草;菅属;叶绿体基因组;系统发育分析;高通量测序;密码子偏好性
菅属Forssk.为禾本科(Gramineae)多年生或一年生草本植物,有30余种[1],其中我国有13种,主要分布于西南和华南地区[2]。黄背草(Willd.) Tanaka是最常见的菅属植物,又被称为进肌草、黄背茅、草糖等,是一种进行C4光合作用的地面芽丛生草[3]。黄背草是非洲、亚洲、澳大利亚热带和亚热带地区生态及经济上最重要和最广泛的多年生草丛之一[4],在全球超过四分之一的草原上占据主导地位[5],其分布几遍中国,多生长于干燥山坡、路边、林缘等处[1]。黄背草在《中华本草》和《全国中草药汇编》中均有记载,其全草均可入药[6],味甘性温,具有活血调经、驱风除湿的功效,主要用于治疗闭经、风湿疼痛[7],具有一定的药用价值。黄背草还具有耐旱耐寒、生长力强、根须繁多等特点[8],其群落通常分布均匀,盖度较大,在一定的范围内可以调节气候、涵养水源[9],具有良好的生态价值。但在药用方面,同为菅属的阿拉伯黄背草Forsk.和中华菅(Linnaeus) Kuntze与黄背草相比不具备相应的价值,且上述3种植物形态学特征极为相似,利用已有的物种鉴定方法难将三者区分开,存在基源植物使用混乱的现象,因此寻找新的方法对其鉴定手段进行补充至为关键。
叶绿体是高等植物和一些藻类所特有的一种半自主性细胞器,也存在于少数原生生物中,具有独立的遗传物质[10]。叶绿体基因组是独立于核基因组的一套完整的DNA序列,结构高度保守且易于测序,其大小介于120~180 kb,共编码120~130个基因[11];叶绿体基因组一般为双链环状分子,大多是以多拷贝的形式存在于细胞中,主要参与光合作用的光反应,在基因表达过程中参与转录和翻译[12]。由于叶绿体基因组编码基因种类和排列顺序稳定,相对分子质量较小,通常可用于植株物种鉴定和分类、系统发育研究及遗传多样性分析等[13]。近年来,随着高通量测序技术的逐渐成熟,越来越多的植物的叶绿体基因组被陆续发布,可见基于叶绿体基因组的研究成为新趋势。在菅属植物中,除黄背草以外的阿拉伯黄背草、中华菅、瘤菅(Nees ex Steud.) Hack.等13种植物的叶绿体基因组也已完成测序[14-16],随着菅属叶绿体基因组数量的增加,同属其他物种的叶绿体基因组也越来越容易组装,这也为黄背草叶绿体基因组的组装提供了条件。目前,国内外大量学者对黄背草的研究多集中在生物学特性[17]、种子萌发[18]和繁殖[19]、生理生态和生产[20]、种群空间分布[21]、对环境因素的生态响应[22]、性状与环境的关系[23]及其作为牧草的适口性和可接受性[24]等方面,然而关于黄背草叶绿体基因组的系统研究尚未见报道,因此,完成黄背草叶绿体基因组图谱的构建和分析,对于后续的物种鉴定以及生物的遗传和进化等一系列研究具有重要意义。
本研究基于Illumina HiSeq高通量测序技术,以黄背草叶片为试验材料,首次对黄背草叶绿体基因组进行测序、组装和注释。同时,将黄背草与其他2种菅属植物叶绿体基因组进行比较,分析了这3种菅属植物的叶绿体基因组特征结构、密码子偏好性、核酸多态性、简单重复序列及其系统发育等信息,进一步促进了黄背草与其他2种菅属植物之间的遗传学和分子鉴定研究,明确了黄背草叶绿体基因组的序列特征、基因组成及系统发育关系,为菅属植物遗传多样性与系统进化研究提供理论依据。
1 材料与方法
1.1 材料
黄背草植物样本采集于河南省沁阳市紫陵镇宋寨村附近太行山系(35°11′33″N,112°45′56″E),由河南科技大学农学院生物科学系洪亚平副教授和戴攀峰博士鉴定为禾本科菅属植物黄背草(Willd.) Tanak,样本保存于河南科技大学标本馆,凭证标本号为TJ-2022101206。部分叶片样本经硅胶干燥后,装入自封袋带回实验室,用无菌水冲洗至表面洁净后置于−80 ℃冰箱保存备用。在NCBI数据库中下载黄背草近缘物种叶绿体基因组序列,试验材料详细信息见表1。
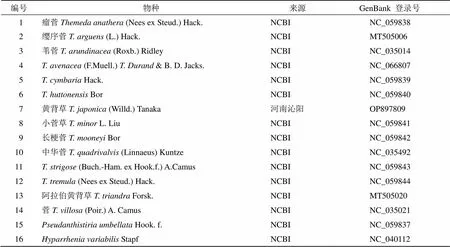
表1 植物样本来源
1.2 叶绿体基因组DNA的提取与高通量测序
取100 mg样本新鲜叶片,液氮研磨后使用植物DNA提取试剂盒(上海联迈生物工程有限公司)进行全基因组DNA提取。将质量浓度大于200 ng/µL且260/280值在1.8~2.0的DNA送至武汉菲沙基因信息有限公司质检,合格的DNA用于文库构建和Illumina HiSeq双端测序,其测序长度为150 bp,去除低质量reads后得到clean reads。
1.3 叶绿体基因组组装与注释
使用SPAdes(v 3.11)软件对上一步得到的clean reads进行基因组组装[25],使用Bandage(v 0.8.1)软件解环并输出完整的叶绿体基因组序列文件[26]。以阿拉伯黄背草叶绿体基因组(GenBank登录号:MT505020)为参考序列,使用在线工具CpGAVAS2(http://47.96.249.172:16019/analyzer/home)对黄背草叶绿体基因组进行注释[27]。注释结果经手动校正后,上传至NCBI数据库,其登录号为OP897809。最后使用在线工具Chloroplot(https://irscope.shinyapps. io/Chloroplot/)绘制其叶绿体基因组圈图[28]。
1.4 叶绿体基因组比较分析
使用IRscope软件对黄背草、阿拉伯黄背草和中华菅(GenBank登录号:NC_035492)叶绿体基因组4个边界的收缩与扩张进行可视化分析[29],并使用在线工具CPJSdraw绘制这3个叶绿体基因组的边界图。使用CodonW(v 1.42)软件对上述3种植物的叶绿体基因组序列进行同义密码子相对使用丰度比较[30],其编码基因按照以下校准进行过滤:(1)基因的起始密码子为ATG;(2)基因长度须大于等于300 bp;(3)处于重复区域的基因只选取1个且去除假基因。使用在线工具mVISTA(https://genome.lbl.gov/vista/mvista/submit.shtml)的Shuffle-LAGAN模式对上述3种植物进行叶绿体基因组比较分析[31]。使用MISA软件对上述3种植物的叶绿体基因组进行简单重复序列分析[32],其参数为单核苷酸SSR:重复单元为10,二核苷酸SSR:重复单元为6,三核苷酸SSR:重复单元为5,四核苷酸、五核苷酸和六核苷酸SSRs:重复单元为4。使用在线软件REPuter(https:// bibiserv.cebitec.uni-bielefeld.de/session Timeout.jsf)对上述3种植物的叶绿体基因组进行长重复序列分析[33],其参数:Hamming Distance为3,最小重复单元为30个碱基。
1.5 黄背草与其近缘物种系统发育分析
使用上述已从NCBI数据库中下载的15条黄背草近缘植物叶绿体基因组序列,以Hook. f.和Stapf作为外群进行全叶绿体基因组系统发育分析。调整序列LSC第一个碱基作为序列起始后,使用MAFFT进行全叶绿体基因组多序列比对,对齐好的文件使用Modeltest软件进行模型检测后,使用RaxML(v 8.2.12)软件在GTR+GAMMA模型下[34],以最大似然法(maximum likelihood,ML)进行系统发育分析,自展值为1000。
2 结果与分析
2.1 黄背草叶绿体基因组结构
原始数据经过过滤后,共得到251 兆条clean reads,且clean reads质量值大于30的碱基占比为88.7%,代表测序质量较好,可用于黄背草的组装。如图1和表2所示,黄背草叶绿体基因组全长为138 736 bp,具有典型的四分体结构,由2个单拷贝区(LSC:84 883 bp,SSC:12 549 bp)和一对重复区(IR:20 652 bp)组成。
在黄背草、阿拉伯黄背草和中华菅序列中,叶绿体基因组长度在138 735~138 961 bp内,阿拉伯黄背草的叶绿体基因组最短。这3种植物叶绿体基因组全长仅相差226 bp,黄背草与其他2种植物IR区域相比收缩了2132 bp,LSC区域扩张了约4000 bp,而SSC区域变化不大。3种植物叶绿体IR区域的GC在43.94%~44.18%,高于LSC区域(36.43%~36.69%)和SSC区域(32.76%~32.78%),这可能是由于IR中含有4个GC含量较高的核糖体RNA(rRNA)基因。
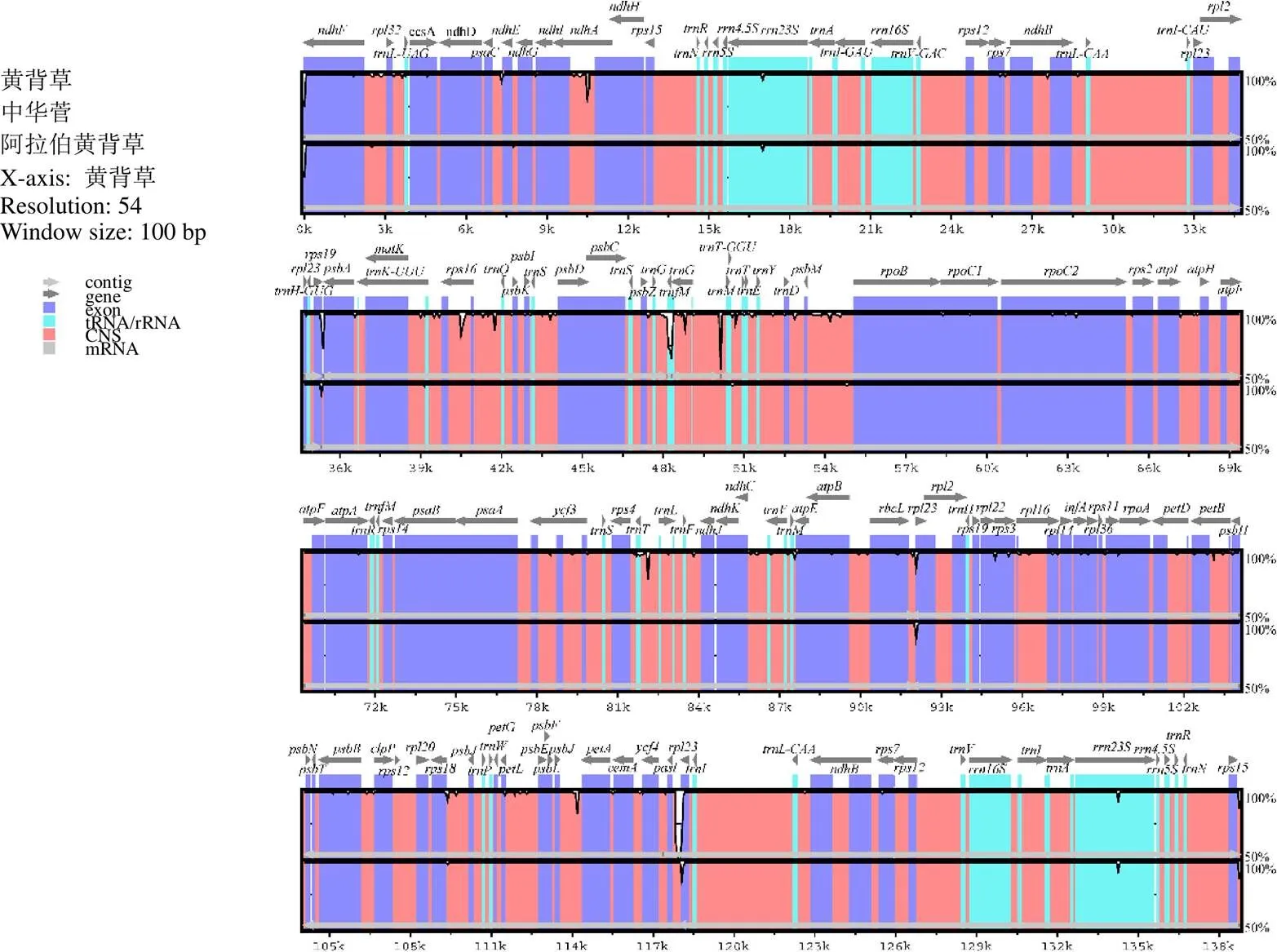
如表3和表4所示,黄背草叶绿体基因组共注释出129个基因,含有82个编码蛋白基因,8个rRNA基因和39个tRNA基因,且IR区域中含有16个重复基因。其中82个编码蛋白基因可以根据不同的功能分为4类:46个光合作用相关基因(photosynthesis related genes),30个自我复制相关基因(self-replication related genes),1个未知功能基因(unknown function genes)和5个其他基因(other genes)。在tRNA基因中,、、、、、、、存在双拷贝,rRNA的4种基因(、、、)存在双拷贝。黄背草IR区注释出16个基因,和阿拉伯黄背草及中华菅相比少了3个基因,分别为、和基因。基因的缺失主要是由于重复区域的收缩引起的,因此本课题组对这3个物种进行了边界分析。由图2可知,黄背草IR区域出现了明显的收缩现象,这导致LSC与IRb及LSC与IRa的边界基因与其他2个物种不同。

外环基因为逆时针转录,内环基因为顺时针转录,相同功能的基因以相同的颜色表示

表2 黄背草、阿拉伯黄背草和中华菅叶绿体基因组碱基组成
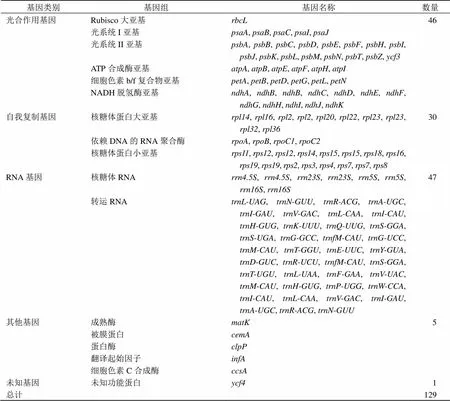
表3 黄背草叶绿体基因组注释基因信息
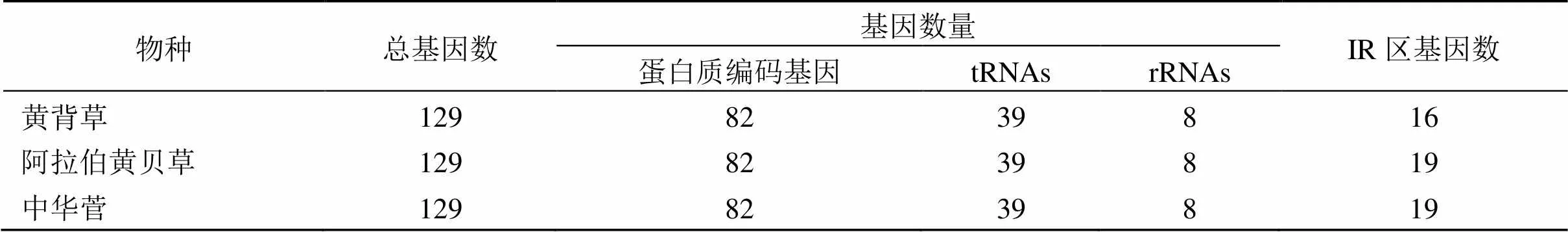
表4 黄背草、阿拉伯黄背草和中华菅叶绿体基因组的基因数及重复基因数目
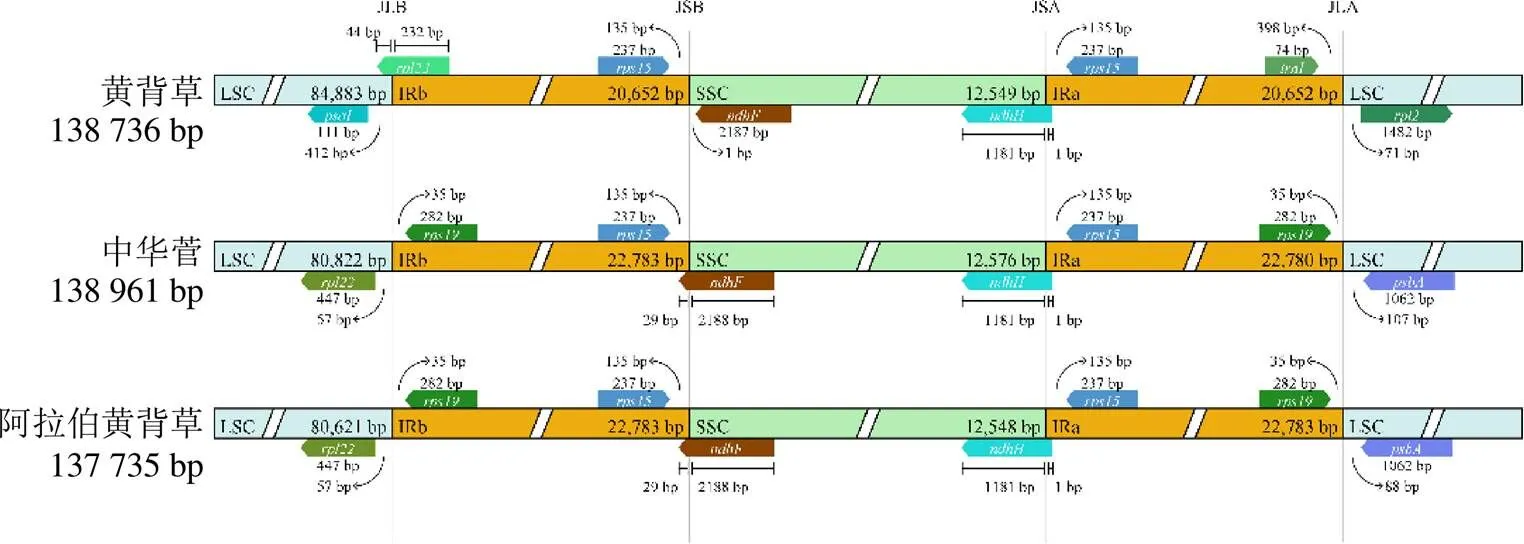
JLB、JSB、JSA、JLA分别代表叶绿体基因组4个边界的连接位点
2.2 密码子偏性分析
按照方法中的3个条件,3种叶绿体基因组经过过滤都只保留了50条蛋白编码序列。其同义密码子相对使用丰度结果见图3,在3种叶绿体基因组中,在三者中相对丰度最大的密码子都为编码亮氨酸(Leu)的UUA;相对丰度最小的密码子在三者中也相同,都为编码亮氨酸Leu的CUG。3种叶绿体基因组的同义密码子相对使用丰度略有不同,但差异不大。由图3可以看出,除了编码色氨酸的(UGG)和甲硫氨酸的(AUG)外,其他氨基酸都有多个密码子。Leu、精氨酸(Arg)和丝氨酸(Ser)有6个同义密码子;甘氨酸(Gly)、缬氨酸(Val)、苏氨酸(Thr)、脯氨酸(Pro)和丙氨酸(Ala)有4个同义密码子;异亮氨酸(Ile)和终止密码子都有3个同义密码子;而其余的氨基酸都有2个同义密码子且同义密码子通常只在密码子第3位不同。
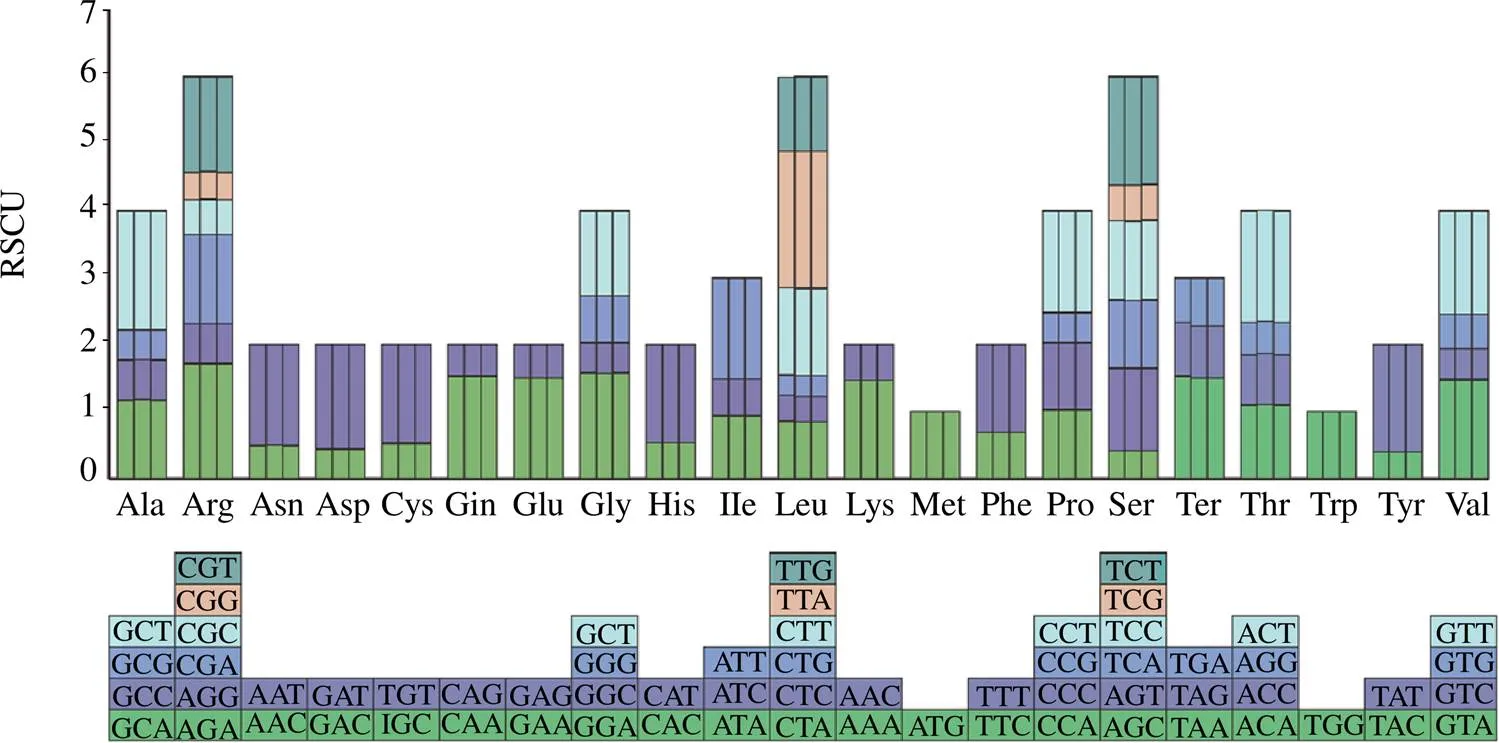
每个氨基酸柱状图从左到右分别为黄背草、阿拉伯黄背草、中华菅
2.3 核酸多态性分析
从3种叶绿体基因组中分别提取到105个共有基因和110个共有间区,经MAFFT对齐后,使用DnaSP 6.0软件计算核酸多态性值(Pi)[35],最后按照基因和基因间区在叶绿体基因组中的位置绘制图谱。由图4可以看出,间区序列的Pi值普遍高于共有基因序列,且IR区域相比于LSC、SSC区域更为保守,这与图5结果相同。在基因序列中,筛选出10个Pi值大于0.001的高突变区域(、、、、、、、、、);在间区序列中,筛选出9个Pi值大于0.003的高突变区域(、、、、、、、、)。突变为进化提供了基础,因此这些高突变区域可用于研究物种进化和物种鉴定。
2.4 简单重复序列(simple sequence repeat,SSR)与长重复序列表征分析
如图6所示,从3个叶绿体基因组中分别检测到38、36、37个SSR。3个叶绿体基因组中都只检测到2种微卫星序列(单核苷酸重复和四核苷酸重复),且大多为单碱基重复型微卫星序列。单核苷酸重复多为A/T重复,其重复单元数目的范围为10~17,四核苷酸重复在三者中都为GTAG重复,中华菅中其重复单元数目为6,而黄背草和阿拉伯黄背草中其重复单元数目为5。SSR广泛分布于叶绿体基因组不同的位置,这些SSR区域可用于物种的分子标记,为遗传图谱构建和目的基因定位提供参考依据。如图7所示,3个叶绿体基因组中都只检测到2种长重复序列(long sequence repeat),包括正向重复序列(forward repeat sequence)和回文重复序列(palindromic repeat sequence),且重复序列长度多为40 bp以下,这些长重复序列可用于研究叶绿体基因组的结构变异。

A-共有基因的核酸多态性分析 B-共有间区基因的核酸多态性分析
横轴代表叶绿体基因组序列长度,纵轴表示序列相似度
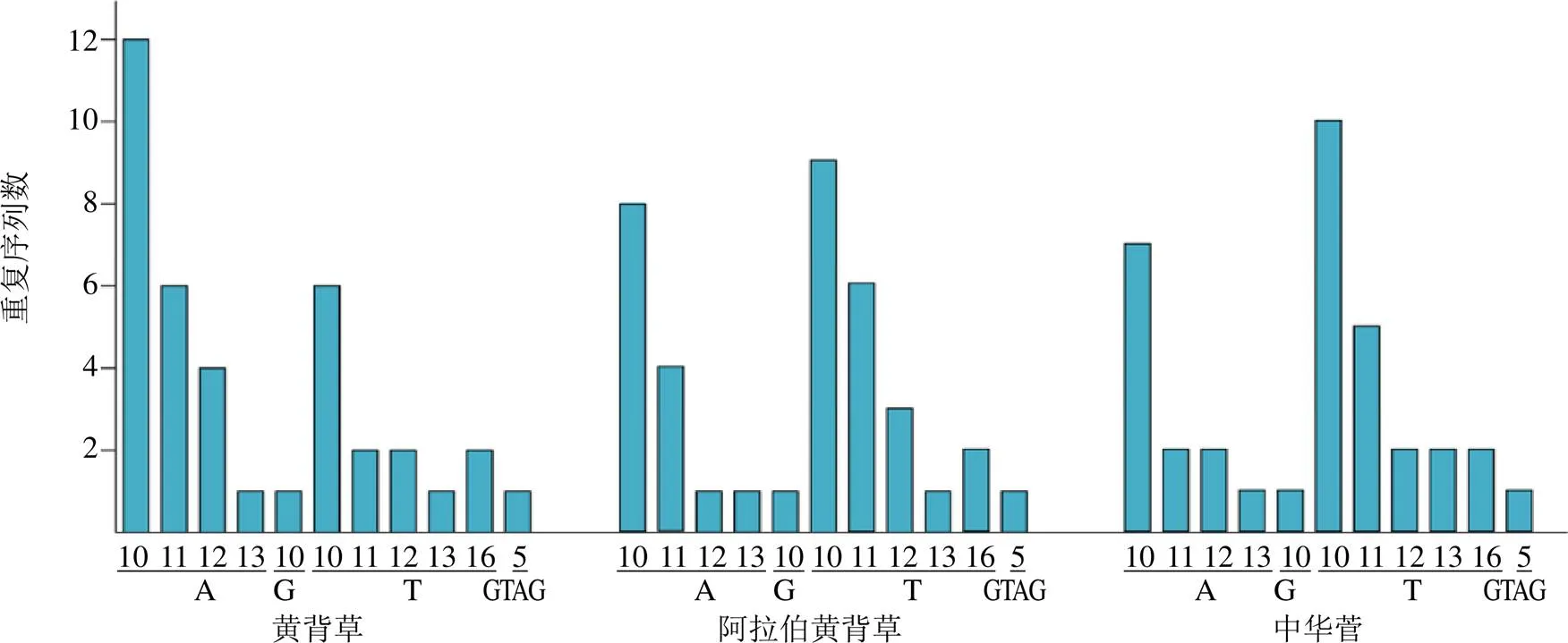
图6 黄背草、阿拉伯黄背草和中华菅叶绿体基因组SSR分析
2.5 系统发育分析
以和作为外群,经Modeltest软件进行模型检测后,使用RaxML(v 8.2.12)软件在GTR+GAMMA模型下[34],以最大似然法(ML)构建黄背草及13种菅属植物叶绿体全基因组的系统发育树(图8)。系统发育分析结果显示,邻接树分支上支持值均大于80%,并具有很好的聚类效果,说明结果的可靠性。所有物种共聚为3大类,分别是外类的和以及菅属,菅属又分为2支。根据进化距离可以看出,相比与禾本科假铁秆草属与苞茅属植物,菅属植物是最后分化出来的一个支系,且黄背草、阿拉伯黄背草和中华菅在菅属植物形成稳定的单系分支,三者中,黄背草与阿拉伯黄背草互为姐妹支,亲缘关系最近。上述结果与《中国植物志》[1]中所记载的分类结果相同。
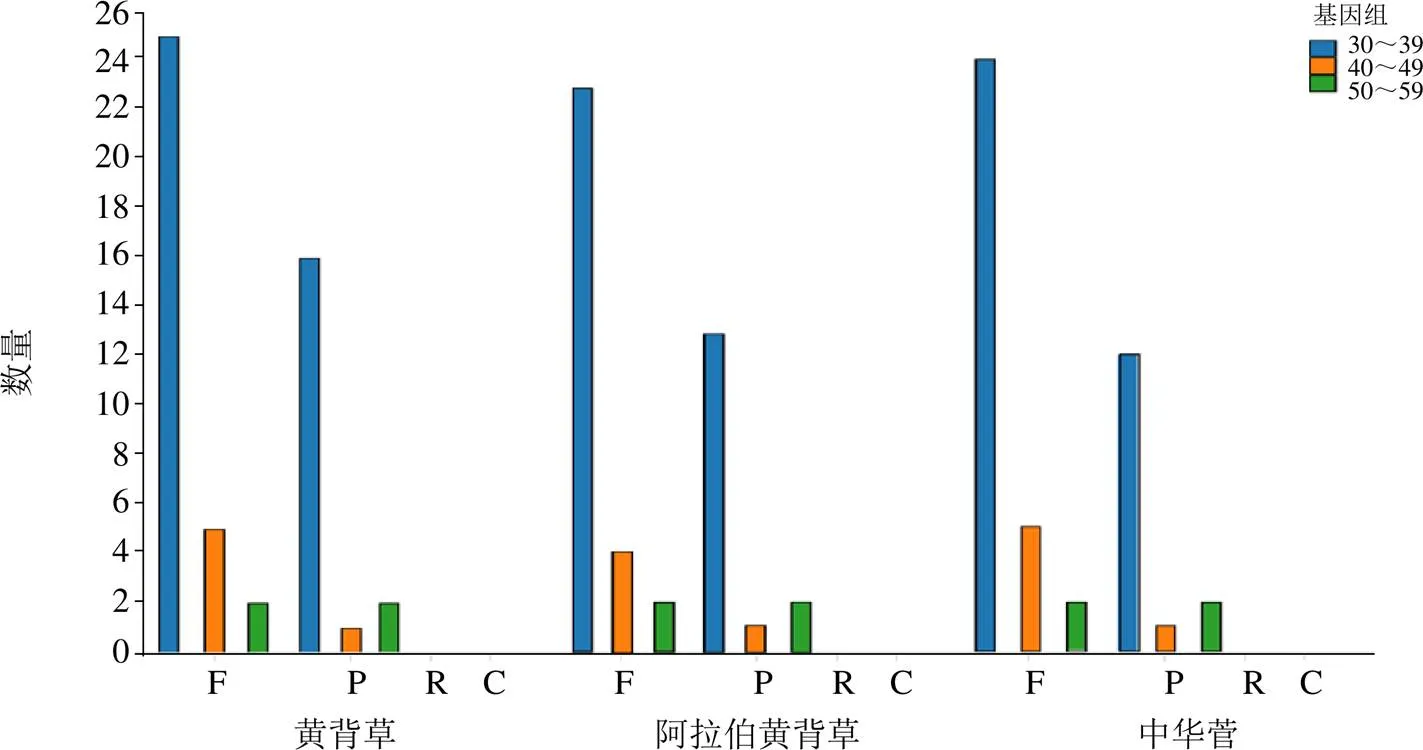
F-正向重复 P-回文重复 R-反向重复 C-互补重复
3 讨论
叶绿体基因组作为独立于核基因组的完整DNA序列,在进化过程中具有高度的保守性和稳定性,近年来广泛应用于植物种类鉴定和系统进化研究[36]。目前已有关于姜属、藜芦属、扁果草属植物中近缘物种叶绿体基因组差异分析及系统发育关系的相关报道[37-38],同时,黄背草与其同属的阿拉伯黄背草、中华菅形态学特征极为相近,以现有的鉴定方法很难区分,这为补充其鉴定方法提供了新思路。
本研究首次完成了黄背草叶绿体基因组的测序、组装和注释工作,将黄背草和已报道的菅属植物阿拉伯黄背草、中华菅的叶绿体基因组进行比较,结果显示其具有典型的四分体结构,3个叶绿体基因组序列全长在138 735~138 961 bp,基因组大小相似度较高,最大仅相差226 bp。同时发现3种菅属植物叶绿体基因组总GC含量(38.53%~38.57%)及LSC区(36.43%~36.69%)、SSC区(32.76%~32.78%)、IR区(43.94%~44.18%)的GC含量也高度相似。对黄背草叶绿体基因组进行注释后得到129个功能基因,其中包括82个蛋白编码基因、39个tRNA基因和8个rRNA基因,与已报道的阿拉伯黄背草、中华菅的叶绿体基因数目一致。黄背草、阿拉伯黄背草和中华菅叶绿体基因组序列的高度相似性在一定程度上解释了三者形态学特征的近似性。IR区被认为是叶绿体基因组中最保守的区域,但生物进化过程中仍经常伴随着IR区的扩张和收缩,这往往会造就叶绿体基因组的差异性[39]。本研究通过边界分析发现,黄背草叶绿体基因组IR区出现了明显的收缩现象,其IR区收缩了2132 bp,LSC区域扩张了约4000 bp,与阿拉伯黄背草及中华菅相比在IR区少了3个基因(、和)。这导致了黄背草LSC/IRb及LSC/IRa的边界基因与其他两个物种的较大差异,而其余2个边界两侧基因完全相同。该差异性可为黄背草物种鉴定及系统发育关系的深入研究提供思路。
密码子使用偏性在解释生物基因组进化规律方面具有重要意义[40]。本研究发现黄背草叶绿体密码子偏好性较低,除了编码色氨酸的和甲硫氨酸外,其他氨基酸都有多个同义密码子,同义密码子之间通常只在第3位存在差异且更倾向于以A/T碱基结尾。同时,黄背草与阿拉伯黄背草、中华菅的叶绿体基因组的同义密码子相对使用丰度具有较高一致性。有研究认为密码子偏性与基因长度、氨基酸组分、GC含量、tRNA丰度等因素有关[40],本研究中3种菅属植物叶绿体基因组的总GC含量及各拷贝区GC含量均低于50%,表明3个叶绿体基因组倾向于使用A/T碱基,这与本实验发现的黄背草密码子更倾向于以A/T碱基结尾这一结论相吻合。本研究从3种叶绿体基因组中共提取到105个共有基因和110个共有间区。经mVISTA工具和DnaSP 6.0软件分析发现,共有间区序列的Pi值普遍高于共有基因序列,且IR区较LSC、SSC区更为保守,这可能是由于IR区的重复基因能够通过基因转换对此区发生的变异进行修正[41]。在基因序列中筛选出10个高突变区域(Pi>0.001),在间区序列中筛选出9个高突变区域(Pi>0.003),这些核酸多态性值较高的变异位点,能够为黄背草物种鉴定的DNA条形码筛选提供参考依据,并且在黄背草及其近缘物种的系统进化研究方面发挥重要作用。
植物叶绿体基因SSR具有数量大、保守性高、遗传信息丰富等特点,其拷贝数的变异是一种重要的分子标记,在植物多态性研究、群体结构分析、种群遗传进化等方面的研究具有重要意义[42]。黄背草、阿拉伯黄背草和中华菅叶绿体基因组中分别检出38、36、37个SSR位点,包括单核苷酸SSR和四核苷酸SRR 2种类型。单核苷酸SSR多为A/T重复,重复单元范围10~17。四核苷酸SSR在三者中均为GTAG重复,其中中华菅重复单元数目为6,其余两者均为5。目前已报道了多种植物叶绿体基因组SSR主要由A/T碱基构成[37-38],这与本实验的研究结果相一致。此外,黄背草、阿拉伯黄背草和中华菅叶绿体基因组中分别检测到51、45、46个长重复序列,且都只检测到正向重复型和回文重复型两种长重复序列,长重复序列是导致叶绿体基因组结构变异的重要因素[43]。这些SSR位点以及长重复序列为黄背草的分子鉴定、物种演化、群体遗传学研究等方面提供了候选分子标记。为进一步确定黄背草在菅属植物中的系统位置,基于黄背草及其15种近缘植物叶绿体全基因组构建ML系统发育树。结果表明,14种菅属植物聚为一类,菅属又分为2支,且黄背草、阿拉伯黄背草和中华菅在菅属植物中形成稳定的单系分支,支持值为100%。其中,黄背草与阿拉伯黄背草互为姐妹支,亲缘关系最为密切。同时,黄背草与阿拉伯黄背草在形态上也较中华菅更为相似,且黄背草与除上述2种植物外的菅属物种相比,形态差异更大,对系统发育分析的结果也起到一定的支撑作用。该研究也表明叶绿体基因组可有效区分菅属植物的系统发育关系,为后续菅属植物的种群分类、系统进化和遗传多样性研究提供材料。
随着高通量DNA测序技术和生物信息学的快速发展,叶绿体基因组因其丰富的遗传信息和高度的保守性,在药用植物的近缘物种鉴定及系统进化研究领域广受关注[36]。本研究首次完成了黄背草叶绿体基因组的序列测序、组装和注释工作,详细介绍了黄背草叶绿体基因组基本特征,阐明了黄背草在菅属植物中的系统位置。同时将黄背草、阿拉伯黄背草和中华菅3种在形态学特征上难以区分的菅属植物的叶绿体基因组进行比较分析,为黄背草的物种鉴定、群体遗传学研究等方面所需的DNA条形码和分子标记筛选提供了参考方向。完成了对菅属植物的遗传多样性与功能基因组学研究的关键补充,为后续开展菅属植物的分子鉴定、系统进化研究以及综合开发利用奠定了理论基础。
利益冲突 所有作者均声明不存在利益冲突
[1] 中国科学院中国植物志编辑委员会. 中国植物志(第九卷. 第二分册)[M]. 北京: 科学出版社, 2002: 23.
[2] 张煜, 刘青. 菅属植物的地理分布 [J]. 热带亚热带植物学报, 2012, 20(3): 221-228.
[3] Christenhusz M J M. The Kew Plant Glossary, an illustrated dictionary of plant terms [J]., 2010, 164(4): 440-441.
[4] Snyman H A, Ingram L J, Kirkman K P.: A keystone grass species [J]., 2013, 30(3): 99-125.
[5] B H, Downing,. Growth and development of fourpopulations from southern Africa in response to temperature [J]., 1985, 51(5): 350-354.
[6] 国家中医药管理局《中华本草》编委会. 中华本草-9 [M]. 上海: 上海科学技术出版社, 1999: 8-429.
[7] 《全国中草药汇编》编写组. 全国中草药汇编(上)[M]. 第2版. 北京: 人民卫生出版社, 1996: 873.
[8] Evans L T, Knox R B. Environmental control of reproduction in[J]., 1969, 17(3): 375.
[9] 赵威, 王馨, 王艳杰, 等. 河南黄背草群落碳、氮密度空间变化及其驱动因素分析 [J]. 草地学报, 2018, 26(4): 846-852.
[10] 热伊汉古丽·图尔迪, 慕丽红, 田新民. 扁果草叶绿体基因组特征分析 [J]. 生物工程学报, 2022, 38(8): 2999-3013.
[11] Tonti-Filippini J, Nevill P G, Dixon K,. What can we do with 1000 plastid genomes? [J]., 2017, 90(4): 808-818.
[12] Khan I A, Zhang Y. Characterization of mango (L.) transcriptome and chloroplast genome [J]., 2014, 85(1): 193-208.
[13] Suparman S, Pancoro A, Hidayat T. Phylogenetic analysis ofbased on rbcL sequences, chloroplast DNA [J]., 2013, 57: 235-240.
[14] Arthan W, Dunning L T, Besnard G,. Complex evolutionary history of two ecologically significant grass Genera,and(Poaceae: Panicoideae: Andropogoneae) [J]., 2021, 196(4): 437-455.
[15] Dunning L T, Liabot A L, Olofsson J K,. The recent and rapid spread of[J]., 2017, 164(4): 327-337.
[16] Dunning L T, Olofsson J K, Papadopulos A S T,. Hybridisation and chloroplast capture between distinctlineages in Australia [J]., 2022, 31(22): 5846-5860.
[17] Tomlinson K, O'Connor T. The effect of defoliation environment on primary growth allocation and secondary tiller recruitment of two bunchgrasses [J]., 2005, 22(1): 29-36.
[18] 赵金辉, 王奎玲, 刘庆华, 等. 黄背草种子萌发特性研究 [J]. 西北农业学报, 2009, 18(3): 245-248.
[19] 岳喜成, 郑观玉, 刘耀曾, 等. 野生黄背草人工繁殖及开发利用试验研究 [J]. 中国水土保持, 1994(12): 29-30.
[20] Ruppert J C, Holm A, Miehe S,. Meta-analysis of ANPP and rain-use efficiency confirms indicative value for degradation and supports non-linear response along precipitation gradients in drylands [J]., 2012, 23(6): 1035-1050.
[21] 王馨, 赵威. 区域尺度下黄背草种群空间分布格局及其与土壤因子的关系 [J]. 草业科学, 2019, 36(2): 335-345.
[22] H A, Snyman,. Transpiration and water-use efficiency in response to water stress inand[J]., 1997, 63(1): 55-59.
[23] Fynn R, Morris C, Ward D,. Trait-environment relations for dominant grasses in South African mesic grassland support a general leaf economic model [J]., 2011, 22(3): 528-540.
[24] van der Westhuizen H, Snyman H, van Rensburg W,. The quantification of grazing capacity from grazing—and production values for forage species in semi-arid grasslands of southern Africa [J]., 2001, 18(1): 43-52.
[25] Prjibelski A, Antipov D, Meleshko D,. Using SPAdes de novo assembler [J]., 2020, 70(1): e102.
[26] Wick R R, Schultz M B, Zobel J,. Bandage: Interactive visualization of de novo genome assemblies [J]., 2015, 31(20): 3350-3352.
[27] Liu C, Shi L C, Zhu Y J,. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences [J]., 2012, 13: 715.
[28] Zheng S Y, Poczai P, Hyvönen J,. Chloroplot: An online program for the versatile plotting of organelle genomes [J]., 2020, 11: 576124.
[29] Amiryousefi A, Hyvönen J, Poczai P. IRscope: An online program to visualize the junction sites of chloroplast genomes [J]., 2018, 34(17): 3030-3031.
[30] Sharp P M, Li W H.usage in regulatory genes indoes not reflect selection for ‘rare’ codons [J]., 1986, 14(19): 7737-7749.
[31] Frazer K A, Pachter L, Poliakov A,. VISTA: Computational tools for comparative genomics [J]., 2004, 32: W273-W279.
[32] Beier S, Thiel T, Münch T,. MISA-web: A web server for microsatellite prediction [J]., 2017, 33(16): 2583-2585.
[33] Kurtz S, Choudhuri J V, Ohlebusch E,. REPuter: The manifold applications of repeat analysis on a genomic scale [J]., 2001, 29(22): 4633-4642.
[34] Stamatakis A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies [J]., 2014, 30(9): 1312-1313.
[35] Rozas J, Ferrer-Mata A, Sánchez-DelBarrio J C,. DnaSP 6: DNA sequence polymorphism analysis of large data sets [J]., 2017, 34(12): 3299-3302.
[36] 姜汶君, 郭梦月, 庞晓慧. 叶绿体基因组在药用植物鉴定及系统进化研究中的应用 [J]. 世界中医药, 2020, 15(5): 702-708.
[37] 马孟莉, 张薇, 孟衡玲, 等. 草果叶绿体基因组特征及系统发育分析 [J]. 中草药, 2021, 52(19): 6023-6031.
[38] 田星, 刘莹莹, 张颖敏, 等. 藜芦属药用植物的叶绿体基因组比较分析和系统发育研究 [J]. 中草药, 2022, 53(4): 1127-1137.
[39] Odintsova M S, Yurina N P. Plastid genomes of higher plants and algae: Structure and functions [J]., 2003, 37(5): 649-662.
[40] 吴宪明, 吴松锋, 任大明, 等. 密码子偏性的分析方法及相关研究进展 [J]. 遗传, 2007, 29(4): 420-426.
[41] Chen J H, Hao Z D, Xu H B,. The complete chloroplast genome sequence of the relict woody plantHu et Cheng [J]., 2015, 6: 447.
[42] 王化坤, 娄晓鸣, 章镇. 叶绿体微卫星在植物种质资源研究中的应用 [J]. 分子植物育种, 2006, 4(S1): 92-98.
[43] Asaf S, Khan A L, Khan M A,. Complete chloroplast genome sequence and comparative analysis of loblolly pine (L.) with related species [J]., 2018, 13(3): e0192966.
Comparison of chloroplast genome characteristics and phylogenetic analysis betweenand two species of
ZHAO Peng-yu1, 2, ZHAO Wei1, HOU Zhi-yang1, LI Xiao1, LIU Bo-kuan1, DU Yao1, XIE Rui-qi3, CAO Ze-lin1, WANG Kai-kai1
1. College of Agriculture, Henan University of Science and Technology, Luoyang 471000, China 2. National Key Laboratory of Plant Molecular Genetics, CAS Center for Excellence in Molecular Plant Sciences, Chinese Academy of Sciences, Shanghai 200032, China 3. College of Basic Medicine and Forensic Medicine, Henan University of Science and Technology, Luoyang 471000, China
was used as the experimental material to compare its chloroplast genome characteristics with those of two congeners,and, and their phylogenetic relationships with the closely related species.Using the Illumina HiSeq high throughput sequencing platform, the chloroplast genome ofwas sequenced for the first time. SPAdes and CpGAVAS2 were used to assemble and annotate it, respectively. CodonW, DnaSP, and MISA were used to perform a series of comparative genomic analysis with two species of plants of the same genus, and the maximum likelihood (ML) method was used to construct a phylogenetic tree.The three chloroplast genomes were 138 735—138 961 bp in length, with a typical tetrad structure, and a total of 129 genes were annotated; compared with the two species of the same genus, the inverted repeats (IR) contracted by 2 132 bp, the large single copy region (LSC) expanded by about 4 000 bp, and the small single copy region (SSC) did not change much. Codon preference analysis showed that the codons with the highest and lowest relative abundance in the three chloroplast genomes were the same, while the relative abundance of synonymous codons was slightly different, but the difference was not significant. Nucleic acid polymorphism analysis showed that the nucleic acid polymorphism values (Pi) of the sequences in the inter-region of the three chloroplast genomes were generally higher than those of the shared gene sequences, and the IR region was more conserved than the LSC and SSC regions. A total of 38, 36 and 37 simple sequence repeats (SSR) and two long sequence repeats were detected in the three chloroplast genomes, respectively. Phylogenetic analysis indicated thatwas most closely related to.The comparative analysis of chloroplast genome characteristics and phylogeny betweenand two species ofwas carried out, laying the foundation for the species identification of the three species and the study of genetic diversity and phylogeny of.
(Willd.) Tanaka;; chloroplast genome; phylogenetic analysis; high-throughput sequencing; codon preference
R286.12
A
0253 - 2670(2023)10 - 3261 - 12
10.7501/j.issn.0253-2670.2023.10.023
2023-02-26
中国科学院大学生创新实践训练计划项目(20224001686);河南省大学生创新创业训练计划项目(202210464086);河南省特色骨干学科建设——旱地绿色智慧农业学科群(17100001);NSFC-河南人才培养联合基金项目(U1304306)
赵鹏宇(2001—),男,在读本科生,主持中国科学院大学生创新实践训练计划项目,研究方向为药用植物分子生物学。E-mail: zhaopengyu122601@163.com
赵 威(1975—),男,教授,研究方向为植物生态学。E-mail: zhaowei1@haust.edu.cn
[责任编辑 时圣明]
