DNA损伤检验点及其在干细胞衰老过程中的调控作用
2023-05-05朱鑫鑫肖建辉
朱鑫鑫,许 艳,肖建辉,2
(1.遵义医科大学附属医院 医药生物技术研究所,贵州 遵义 563000;2.遵义医科大学附属医院 妇产科,贵州 遵义 563000)
细胞衰老最初被定义为细胞在体外经过有限次的分裂后,导致端粒磨损,从而触发了以DNA损伤为信号的细胞周期阻滞[1]。DNA损伤在分子水平上的影响包括基因组不稳定、端粒功能障碍、表观遗传改变、蛋白质应激和线粒体功能受损等,所以DNA损伤可以说是导致干细胞衰老的驱动因素[2-5]。研究表明,衰老干细胞相比年轻干细胞积累了更多的DNA损伤,而正常的细胞衰老过程中,由于大量外源性因素(辐射、紫外线、毒性化学物质等)和内源性基因毒素,DNA损伤会持续大规模的发生,引起DNA单/双链断裂以及细胞内DNA损伤标志物γH2AX的产生,扰乱细胞的稳态平衡,导致细胞功能异常包括凋亡、过早分化以及衰老[6]。针对DNA损伤导致的严重伤害,生物体进化出高度保守的精密程序:DNA损伤检查点 (DNA damage checkpoint),保证细胞周期中DNA复制和染色体分配质量,帮助协调DNA复制和损伤清除[7]。根据检验点存在的时相不同,可分为G1期末检验点、S期内检验点、G2期末检验点以及纺锤体组装检验点等[8]。在DNA损伤过程中,DNA损伤应答和多种细胞周期检验点调控因子被启动,并通过一系列磷酸化级联反应,减缓细胞周期进程,为DNA修复提供充足的时间,若修复无望则直接进入细胞程序性死亡,从而保证基因的完整性遗传。不同类型的干细胞具有不同的DNA损伤反应(DNA damage response,DDR)机制,而与周期检验点相关调控因子的变化会导致干细胞衰老,主要表现在干细胞数量减少、分化能力降低以及表观遗传改变等。成体干细胞的衰老和耗竭,使生命体受损组织器官丧失了自我修复能力,引起与年龄相关疾病的发生发展。研究DNA损伤检验点中调控因子对干细胞衰老的作用,是阐明DNA损伤诱导的衰老机制的关键。因此,本文将对DNA损伤检验点的工作模式以及相关调控因子对干细胞衰老过程中的作用进行综述。
1 DNA损伤检验点及其工作模式
DNA损伤检验点属于细胞周期检验点的一类,是细胞内存在的一种监控机制,可以识别细胞周期中出现的DNA损伤以及结构异常,并诱导产生特异的抑制因子,阻止细胞周期进程[9]。在DNA发生损伤后,检验点ATM/ATR-CHKl /CHK2-cdc25A/B/C通路的成员相继被激活亦或失活,最终由CDK-cyclin复合物感受信号,决定细胞周期的进程以及细胞命运[10]。DNA损伤检验点通过调控细胞周期的3个阶段来监控DNA的损伤情况:G1/S期的转换、S期整个进程以及G2/M期临界点,以保证基因和基因组的稳定性(图1)。
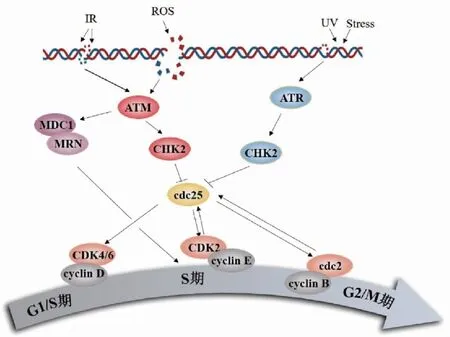
图1 细胞周期检验点
1.1 G1/S期检验点 正常情况下,真核细胞由G1期进入S期,开始DNA合成并继续运行细胞周期,直至完成细胞分裂。但如果此时出现DNA损伤,G1/S期检验点阻止DNA复制过程的起始,以阻止受损DNA进行复制,从而使细胞周期不能进入S期。ATM和ATR作为DNA损伤的感应分子,在不同程度的DNA损伤中发挥各自的作用。当G1期细胞受到电离辐射或其他引起DNA双键断裂的化合物处理致使DNA损伤时,ATM被激活并通过磷酸化细胞周期检测点激酶1 (checkpoint kinase 2,CHK2)的Thr68位点,进而介导细胞分裂周期-25A(cell division cycle 25 A,cdc25A)上Ser123位点的磷酸化,通过与14-3-3蛋白结合从核内转运出来,并进一步被泛素化降解失活[11]。cdc25A蛋白的失活阻止了CDK2的磷酸化,导致DNA复制的短暂阻断,细胞周期阻滞在G1期。若DNA损伤是由紫外线照射或其他引起DNA单链损伤的因素造成的,ATR则是主要的感受器,通过中介因子TOPBP1、claspin和9-1-1(Rad9-rad1-hus1)复合物等介导,随后细胞周期检测点激酶1 (checkpoint kinase 1,CHK1)被激活,进而磷酸化cdc25A,导致G1期阻滞[12]。
1.2 S期检验点 当S期阶段发生DNA的损伤或未修复的损伤转化为继发性损伤时,S期检验点被激活,从而抑制复制起点的启动,使DNA复制速度减慢,同时激活DNA修复和复制叉的恢复等机制,协调细胞周期的正常进行,确保细胞进入有丝分裂前DNA的完整性和精确性。
S期检验点主要通过两条信号通路来发挥作用,包括经典的ATM/ATR-CHK2/CHK1-cdc25A-CDK2通路和染色体结构维持蛋白1 (structural maintenance of chromosome protein 1,SMC1)的磷酸化,主要作用是通过抑制复制起始点,引起S期的阻滞。当UV或化学物质引起DNA单链损伤时,在中介因子TOPBP1和9-1-1的作用下,ATR结合到单链DNA上而活化[13]。随后,复制叉相关蛋白Claspin将CHK1招募到停滞的复制叉处,并被ATR磷酸化激活,活化的CHK1磷酸化其底物cdc25,从而抑制cyclin E/A-CDK2的活性[14]。cyclin E/A-CDK2活性受到抑制后,阻止未在起始复制的复制起点上的cdc45的募集,以此来阻止DNA复制启动[15-16]。已在复制叉位点上的cdc45和DNA解旋酶(mini chromosome maintenance protein,MCM)则以独立于CDK2的方式相互作用,负调控复制起点CHK1的激活[17]。另外,Claspin 还可通过在S期DNA的复制启动过程中与cdc7相互作用促进MCM蛋白磷酸化[18]。另一条通路则是通过染色体结构维持蛋白SMC1的磷酸化实现S期的延长。SMC1的磷酸化依赖于ATM-MDC1-MRNC(Mrc11-Rad50-Nbs1 complex)等中间产物的激活,从而发挥染色体修复功能。在辐射刺激下,激活的ATM被DNA损伤检查点1的中介物(mediator of DNA damage checkpoint 1,MDC1)和MRNC招募到断裂的位置,磷酸化SMC1上的Ser360和Ser957位点,导致S期检验点缺陷,DNA复制停滞[19]。
1.3 G2/M期检验点 G2/M检查点阻止DNA受损的细胞进行有丝分裂,从细胞周期中期过渡到后期。有丝分裂激酶CDK1作为G2/M期转换的关键蛋白,通过使底物蛋白磷酸化,改变其下游靶蛋白的结构和启动其功能,实现其调控细胞周期的作用,其活性受到cdc2上Tyr15和Thr14的抑制性调节[20]。研究表明,CDK1在细胞周期进程中通过G2/M相变和同源重组修复通路的激活发挥关键作用,抑制CDK1的活性可逆转DNA损伤,调控细胞周期;而cdc2基因突变,CDK1活性增强,则会导致细胞周期阻滞在G2/M期[21]。此外,在CDK1缺陷型小鼠中过表达cyclin A使CDK-cyclin A过度活化,上调ATR/CHK1导致细胞周期阻滞在G2/M期[22]。可见CDK1是调节细胞周期中期和后期转换的关键检验点。在有丝分裂中,p70S6K是CDK1的直接底物,当CDK1活性较高时,S6K1在多个Ser/Thr位点发生磷酸化或去磷酸化,导致S6K1失活,致使核蛋白的生成,帮助细胞更好的通过有丝分裂[23]。此外,p70S6K也受哺乳动物雷帕霉素靶点(mammalian target of rapamycin, mTOR)的调控,在DNA损伤情况下, mTOR通过磷酸化下游p70S6K阻滞细胞周期向后期转化[23];当mTOR敲除后,CHK1总蛋白以及磷酸化减少,G2/M细胞周期阻滞被缓解。
2 DNA损伤检验点对干细胞衰老过程的调控作用
DNA损伤检查点不仅影响干细胞对DNA损伤的反应,而且对干细胞衰老相关生物学过程起重要调控作用,包括自我更新、分化、周期阻滞以及凋亡等(图2)。研究表明,在长期培养过程中,端粒长度缩短以及外源因素如氧化应激均会导致DNA损伤[24],增加γH2AX病灶的形成和DNA甲基化等,导致干细胞的衰老。而DNA损伤修复相关基因的缺乏或下调,会导致干细胞的衰老及其功能的失调[25-26]。可见检验点和干细胞衰老过程中相关功能的变化之间存在着密切的联系,剖析DNA损伤检验点调控干细胞衰老进程的分子机制有助于深入探究干细胞衰老。以下简述了DNA损伤检验点相关信号对干细胞衰老相关生物学过程的调控作用。

图2 细胞周期检验点调控干细胞衰老过程中的生物学过程
2.1 ATR/ATM-CHK1/CHK2 ATR/ATM作为上游检查点起始激活组件,是维持干细胞的稳态的第一道关卡。ATR和ATM调节DNA损伤中p53的激活,两种激酶的缺失都会导致干细胞快速耗竭[27]。研究表明,ATR检验点激酶的缺陷,会导致干细胞更新能力的降低和组织稳态失衡,DNA损伤修复能力下降以及与衰老表型的过早出现。而下游CHK1和p16的表达随着年龄的增长而增加,导致老年干细胞相比于年轻干细胞,更易受DNA损伤的影响[28]。ATM检验点的缺失也会加重DNA损伤积累并加速组织衰老[26]。我们最近的研究也发现,在DNA损伤药物和ATR抑制剂共同处理后的干细胞,其细胞衰老程度显著增加,表现为β-半乳糖苷酶阳性细胞增多,DNA损伤标志物表达增加,细胞活力下降。
在端粒功能障碍的小鼠中,ATR的活性可被核酸外切酶(exonuclease 1,Exo1)间接调控,Exo1位于端粒末端,通过DNA末端切除在功能失调的端粒诱导产生DNA损伤信号,从而磷酸化ATR和CHK1产生单链DNA,激活DNA 损伤检查点[29],改善辐射诱导的干细胞周期阻滞和DNA损伤诱导的组织衰老[30]。此外,在端粒功能障碍的哺乳动物中,Exo1的缺失降低了ATR活性以及p53的生成,导致细胞周期阻滞和凋亡的减少,维持了干细胞和组织的功能并延长了哺乳动物的寿命[30]。而ATR/ATM下游信号CHK1/CHK2作为细胞周期的关键调节器,抑制CHK1会引起造血干/祖细胞(hematopoietic stem and progenitor cell,HSPC)DNA 损伤的累积并停止分裂,导致造血功能障碍[31]。作为DDR的下游效应因子,CHK2的缺失可以挽救端粒酶缺陷小鼠的寿命和年龄相关的病理,而在DNA损伤诱导下,CHK2的激活延迟干细胞的自我更新和分化,以防止组织过早老化和癌干细胞的形成[32]。此外,ATR/ATM途径对肿瘤干细胞DNA损伤修复有重要调控作用。肿瘤干细胞由于DNA损伤应答反应存在缺陷,更依赖ATR/ATM修复途径,因此对其抑制剂更为敏感。当其中一个途径发生突变导致缺陷,抑制互补通路就会对肿瘤细胞造成“合成致死”。通过ATR/ATM-CHK1/CHK2抑制剂的开发,增强肿瘤细胞以及肿瘤干细胞对DNA 损伤的敏感性的治疗方式可能是一个良好的策略[33]。
2.2 p53-p21 p53-p21信号作为细胞周期检验点相关基因在干细胞衰老相关生物学过程中发挥重要作用,包括细胞周期、自我更新、分化以及凋亡。
p53作为一个抑癌基因被发现,同时也是一种重要的转录因子,当p53发生突变,细胞增殖的负调控作用丧失,则导致细胞周期失控而过度增殖[34]。p53的缺失可增强小鼠造血干细胞的自我更新能力[35]。同样,作为p53的下游基因,p21的缺失也会增强造血干细胞的增殖能力,导致过量的造血干细胞进入周期增殖,破坏了造血干细胞的相对静止状态,导致干细胞过早耗竭以及功能丧失[36]。p21除了受p53调控外,也可通过非依赖p53的方式被激活,包括p53家族成员 p73和CHK2。在p53敲除的干细胞中,p21表达量上调,DNA 修复反应被激活,并保持自我更新能力,恢复了受损组织的能力[37]。此外,p53基因的缺失可以挽救表皮干细胞功能,促进皮肤更新和创面愈合反应[38]。有趣的是,p53对于端粒功能障碍的干细胞则会产生不同的作用。在端粒异常的肠上皮干细胞中,p53的缺失影响肠干上皮细胞染色体的稳定性,导致干细胞分化缺陷和肠道早衰[39]。与组织干细胞不同,胚胎干细胞在发生DNA损伤后较少经历依赖p53的细胞周期阻滞。Jaiswal等[40]的最新研究显示,在胚胎干细胞进入分化之前受到DNA 损伤诱导应激后,以p53非依赖的方式激活干细胞DNA损伤检验点和凋亡;而当胚胎干细胞进入分化期后,p53则通过维持DNA甲基化的平衡,增加基因组稳定性以此来促进干细胞的分化[41]。p53能够维持干细胞的静止状态,则其缺失可能会使干细胞对辐射诱导敏感。在应对DNA损伤反应中,活化的p53通过激活下游蛋白p21,通过与CDKs和cyclin复合物相结合以延缓细胞周期,为DNA修复提供时间。而p53在决定细胞命运中的作用通常由细胞所经历的确切损伤水平来调节的。
2.3 p16-pRb p16是一种细胞周期蛋白依赖性激酶抑制剂,通过Rb检查点功能来稳定细胞周期阻滞[42]。Rb蛋白是G1期的关键调控因子,也是转录因子E2F的抑制因子,Rb的过度磷酸化导致E2F从E2F/pRb复合物中分离,并促进G1/S过渡,而E2F/pRb作为G1期向S期转变的关键调节信号通路已被证明与ESCs的分化和自我更新有关[43-44]。
p16在年轻组织中表达水平极低,并随着年龄的增长积累,调节HSC的功能。在缺失p16的情况下,HSC在增殖缺陷和细胞凋亡方面得到缓解,表明低水平的p16表达可能有助于维持干细胞中的HSC静止,缓解HSC衰老枯竭[45],并增强小鼠衰老干细胞(如HSC和神经干细胞)和再生组织(如胰岛)的功能[46-47]。虽然p16介导的干细胞衰老的机制尚不完全清楚,但是p16阳性细胞数量随着年龄增加而增加,并对寿命产生负面影响。从这个角度出发,清除p16阳性细胞可能有助于治疗与年龄相关的功能衰退[48]。有报道显示,在Rb1敲除小鼠中,成骨干细胞中pRb的表达被有效地消除。这些动物在骨骼上表现出明显的发育缺陷,最明显的是颅骨[49]。另外,在Rb缺陷的小鼠中,卫星细胞数量在6个月增加了5倍,此外,成肌细胞也增加了3倍,而终端分化大大减少。Rb的缺陷加速了卫星细胞从静止状态再进入细胞周期活跃状态,促进肌肉中卫星细胞的分化。相反,持续的Rb1缺失会导致肌纤维形成缺失[50]。通过响应DNA损伤和其他形式的细胞应激而激活的p16,可能会对干细胞的功能产生不利影响,而P16在DNA损伤驱动衰老中的作用尚未在DNA损伤或端粒功能障碍的小鼠模型中单独研究。因此,研究这些基因在这些条件下的功能具有特别的意义。
2.4 CDKs-cyclin CDK是丝氨酸/苏氨酸激酶,其催化活性受cyclin和 CDK抑制因子(cyclin dependent kinase inhibitor, CDKI)相互作用的调节。哺乳动物CDKs和cyclin在细胞周期的每个阶段都会改变,确保以有序的方式完成细胞周期进程,满足复杂多细胞生物进化过程中对不同细胞类型增殖的更精细控制的要求[45]。目前,CDKs、cyclin和CKI家族成员涉及到转录、DNA损伤修复、蛋白酶降解、表观遗传调控、代谢、干细胞自我更新等多种生物过程[51]。
作为细胞中细胞周期调节因子如cyclin、CDKs以及CKI通常以转录因子为靶点,引发转录程序的全局变化。G1期cyclin及其相关的CDKs可以磷酸化并稳定核心多能性转录因子Nanog、Sox2和Oct4,缺失G1细胞周期蛋白的情况下,细胞虽然可以继续增殖,但是随着周期蛋白的消融,胚胎干细胞的多能性降低[52]。敲除CDK1、CDK2、cyclin E或B1以及抑制CDKs的活性均会导致干细胞多能性的丧失。CDK1作为Oct4的上游因子,可通过影响Aurkb/pp1信号通路协调Oct4的活化并与染色体结合,进而把细胞周期和多能性联系起来[53-54]。此外,CDK1激酶也可通过调节 PI3K/Akt 通路来维持干细胞多能状态[53]。因此,调节G1期CDKs以及cyclin可能是调控干细胞多能性命运的一个关键。
除周期蛋白和调节因子对CDK活性进行调控之外,细胞内还存在一些对CDK活性起负调控的蛋白质,称为CDKI,包括 Cip/Kip家族的p21,p27和p57,能够阻断cyclin E-CDK2和cyclin A-CDK1的活性,以及INK4家族的蛋白, p16、p15、p18和p19抑制cyclin D-CDK4/6的活性[55]。在我们目前的研究中也发现,p21、p16表达下调,相应的CDKs,如CDK1、CDK2的表达量上升,细胞周期阻滞情况得到改善,干细胞呈现年轻化。除此之外,有研究表明,p21和 p27与 Sox2增强子的结合可以启动神经干细胞和胚胎干细胞的分化[56-57]。而p21缺失后,过表达Sox2则会诱导干细胞复制应激和DNA损伤反应,导致细胞生长停滞[57]。这些调节因子通过调控细胞周期阻滞,促进干细胞的程序性凋亡,对调节组织稳态和预防肿瘤发生具有重要意义。
3 结语
ATM和ATR作为DNA损伤检验点的核心部分,在不同的DNA受损条件下被招募到损伤部位,通过磷酸化和去磷酸化CHK1/CHK2和cdc25家族引发一系列级联反应,以应对DNA损伤的发展,进而调控细胞生理进程。DNA损伤检验点在干细胞衰老相关生物学过程中发挥着重要的作用,包括调控干细胞的自我更新、分化、周期阻滞以及凋亡,其中涉及了多条信号通路的参与,包括p53/p21、p16/pRb以及Exo1基因等。探究由DNA损伤检验点调控干细胞衰老的分子机制是重要的,这对于探究如何维持干细胞的功能有着关键的作用。然而,DNA损伤检验点如何通过影响干细胞的功能,进而影响机体,以及检验点和其他信号通路之间存在的交互作用还需要通过体内和体外研究进行深入分析,这也是我们接下来研究的重点。在自发或外源因素刺激干细胞发生DNA损伤后,除了激活DNA损伤检验点以保证基因完整性遗传外,是否涉及其他机制的变化,以及两者之间是否存在联系,都是待解决的问题。揭示 DNA 损伤和衰老之间关系,为进一步探索DNA损伤在衰老病理发展过程中的机制,从根源上解决衰老过程提供了方向,同时为应对衰老相关的疾病提供了新的思路。
