钯基催化剂氧化低浓度甲烷的研究进展
2023-05-05赵旭腾王一男张毅然
赵旭腾,侯 进,王一男,桂 煜,张毅然,李 珂,陈 婷,林 赫, *
(1. 上海交通大学 机械与动力工程学院, 上海 200240; 2. 中国船舶集团有限公司第七一一研究所, 上海 200090)
0 引 言
国际航运温室气体排放控制是全球温室气体减排的重要组成部分,也是全球船舶行业共同面临的责任和挑战。2018年国际海事组织(IMO)提出了温室气体减排战略(MEPC 72),计划到2030年和2050年,船舶碳强度相对2008年分别降低至少40%和70%。低碳燃料的高效利用是减少环境污染、降低温室气体排放的重要方式。以甲烷(CH4)为主要成分的天然气因资源丰富、应用技术成熟而受到广泛关注。2021年,全球液化天然气(LNG)运输市场活跃船舶达到642艘,全球LNG海运量超3.7亿t;2021年全球共新增订购240 艘LNG动力船,签订的LNG 动力船舶的新造船合同数量超过去四年订购数量总和。与传统燃油动力相比,使用LNG可以在整个产业链周期内立即实现20%以上碳减排,同时降低85%以上NOx排放以及99%的SOx排放。在H2、NH3等“零碳”燃料难以短期内安全、高效应用的情况下,天然气是目前满足全球航运业温室气体减排目标唯一可行的大规模航运燃料解决方案。
然而,CH4的温室效应是CO2的25倍[1],天然气发动机缸内燃烧不充分导致的未燃CH4(浓度大约0.05%~0.5%)逃逸已成为大气中CH4最主要的来源之一。据国际清洁交通委员会统计,2012—2018年全球船舶排放的CH4增加了150%,未燃CH4的过量排放会抵消天然气作为替代燃料的碳减排优势,甚至造成更严重的温室效应。我国颁布的《船舶发动机排气污染物排放限值及测量方法(中国第一、二阶段)》中已经限定了第1类和第2类天然气或天然气双燃料船舶发动机的CH4排放。可以预见,未来船用双燃料/气体发动机CH4排放法规势必将在全球范围内逐步建立和完善。
1 低浓度甲烷催化氧化技术
天然气发动机排气中的CH4泄露来源于燃料燃烧不充分,催化氧化技术是目前消除尾气中未然甲烷的主要方式。催化氧化技术的核心在于获得适用于目标反应的高活性、高可靠性催化剂[2]通常面临以下技术挑战[3-5]:
(1)催化剂低温活性问题。CH4具有稳定的四面体结构,化学稳定性极高导致活化困难[6]。在达到可燃浓度范围(5%~15%)并且氧气充足的情况下,直接燃烧温度需达到800 ℃,而天然气发动机排气中CH4浓度仅有0.05%~0.5%,无法直接燃烧去除。而船用低速和中速发动机排气温度分别在200~300 ℃和300~400 ℃范围内,远低于CH4的正常起燃温度。因此,开发低温高效CH4氧化催化剂是首要问题。
(2)恶劣工况条件下催化剂可靠性问题。天然气发动机排气中包含着CO、SOx/H2S、非甲烷碳氢NMHC和水蒸气等其他气体组分,会对CH4催化燃烧效率产生不利影响,甚至导致催化剂失活。因此,在提高催化剂低温性能的同时,还需要保证催化剂能在复杂恶劣的尾气氛围中长期稳定运行,避免催化剂活性组分烧结或中毒失活[6]。
(3)催化剂研发成本昂贵问题。钯(Pd)对于CH4有着高效的催化氧化性能,但同时Pd也是世界上最稀有的贵金属之一。全世界Pd的年总产量不到黄金的8%,其价格走势与供求之间存在非常紧密的相关性。近年来,随着排放法规的日益严苛,全球汽车、船舶等工业对Pd的需求量不断增大,而供给方面却凸显不足,导致Pd的价格大幅上涨,甚至超越黄金成为最昂贵的贵金属。因此,如何在提升催化剂性能的同时,尽可能减少催化剂中贵金属的用量以降低成本是催化剂研发重点。
2 钯基甲烷氧化催化剂简介
美国福特公司于1989年首次研发出Pd/Rh催化剂[7],与Pt、Rh等其他贵金属催化剂相比,Pd基催化剂不仅经济性高,耐热烧结性优异,而且对碳氢化合物有着更好的氧化效率[8-9]。考虑到实际工况条件,Pd基催化剂的研究重点集中在解决CH4的低温起燃、H2O的抑制失活、CO或SOx中毒、高温稳定性和耐久性以及成本等问题[10-18]。除了催化剂整体的比表面积和孔结构以外,许多因素会深刻影响Pd基催化剂的催化性能,例如,Pd物种分散状态及暴露晶面、Pd粒径尺寸、Pd价态分布、PdO与金属Pd之间的相变、载体种类、金属-载体间相互作用(SMI)程度以及Pd与其他组分间的协同效应等[19-23]。针对这些基础科学问题开展研究将有助于深刻理解Pd活性位在CH4氧化过程中的运作机制,明晰不同组分间的相互作用,为未来开发更多低温高效、稳定性高的Pd基催化剂提供理论指导。接下来,将分别针对影响Pd基催化剂的关键因素,Pd基催化剂的甲烷氧化机理以及Pd基催化剂的失活机制等方面的研究进行详细介绍。
3 钯基催化剂的构效关系研究
3.1 Pd粒径分布对催化活性的影响
Pd粒径尺寸变化会影响暴露晶面、不饱和位点的数量并改变Pd与O之间的键能,体现出结构敏感性[24]。Hicks等[25]最早探索了Pd/Al2O3催化剂表面Pd颗粒尺寸对CH4催化氧化过程的影响。研究表明,相比于Pd粒径为1.4 nm的催化剂,粒径为4.0 nm左右Pd颗粒的单位活性位点转化频率(turnover frequency,TOF)更高,由0.02 s-1提升至1.3 s-1,但表观活化能几乎一致。Stakheev等[26]发现在稀燃条件下Pd粒径从1 nm增加到20 nm时,催化剂TOF线性增加,进一步证实了富氧条件下CH4氧化在Pd粒径方面的结构敏感性,粒径大的PdO颗粒催化性能更好。大多数研究者认为这是由于相对较小(1~2 nm)的PdO颗粒拥有更强键能的Pd—O键,限制了CH4中C—H键活化。然而,PdO粒径的过度生长也会导致Pd分散度降低,不利于Pd的高效利用。因此,有研究者倾向于认为中等尺寸(~4 nm)的PdO纳米颗粒具有最高的TOF。Christian等[27]通过改变预还原温度控制Pd的粒径和价态,研究了富氧条件下CH4在Pd/ZrO2催化剂上的氧化过程。结果显示,随着Pd粒径增大,Pd颗粒与ZrO2的接触界面变小。尽管与载体之间的相互作用减弱,但有利于CH4氧化性能提升。
反应氛围、载体种类以及与载体之间相互作用也会影响催化活性与Pd粒径之间的构效关系。Chen等[28]研究了化学计量反应条件下Pd/Al-Ce催化剂表面PdO纳米颗粒在CH4氧化过程中的粒径效应,发现受Pd-Ce间相互作用的影响,粒径小的PdO颗粒有着更高的CH4转化率。结果表明,化学计量反应条件下1.9 nm的PdO颗粒表现出更强的Pd-Ce相互作用,更容易被还原为高活性的金属态Pd0,实现CH4转化率的大幅提高;而较大的8.5 nm的PdO颗粒与载体CeO2之间的相互作用较弱,呈现了较低的单位活性位点TOF和较高的活化势垒。因此,在不同的富燃/稀燃条件下,Pd粒径的改变可能会影响Pd0-Ce和Pd0-PdO界面之间的相互作用,影响催化活性。
Cargnello等[20]发现不同载体(Al2O3、 MgO、 SiO2、 Ce0.8Zr0.2O2)上CH4氧化活性对于均匀的Pd颗粒(2~9 nm)尺寸依赖性较弱,并将这种结构不敏感性归因于不同载体上PdO(101)和(100)晶面比例的变化。Murata等[29]的研究表明,不同粒径Pd颗粒(1~20 nm)在α/θ-Al2O3上的CH4氧化活性呈火山形曲线,而Pd在γ-Al2O3上的尺寸依赖性单调增加。这种尺寸依赖性可归因于Pd粒径以及Pd与载体相互作用改变而引起的颗粒表面台阶活性位点比例的变化,如图5(b)所示。综上所述,Pd粒径是影响CH4性能的关键因素,但不同的进气氛围、载体种类等会影响不同粒径Pd与其他组分间的相互作用方式和程度,因此,Pd粒径对于CH4氧化过程的影响也存在显著差异。
3.2 Pd分散态及价态分布对催化活性的影响
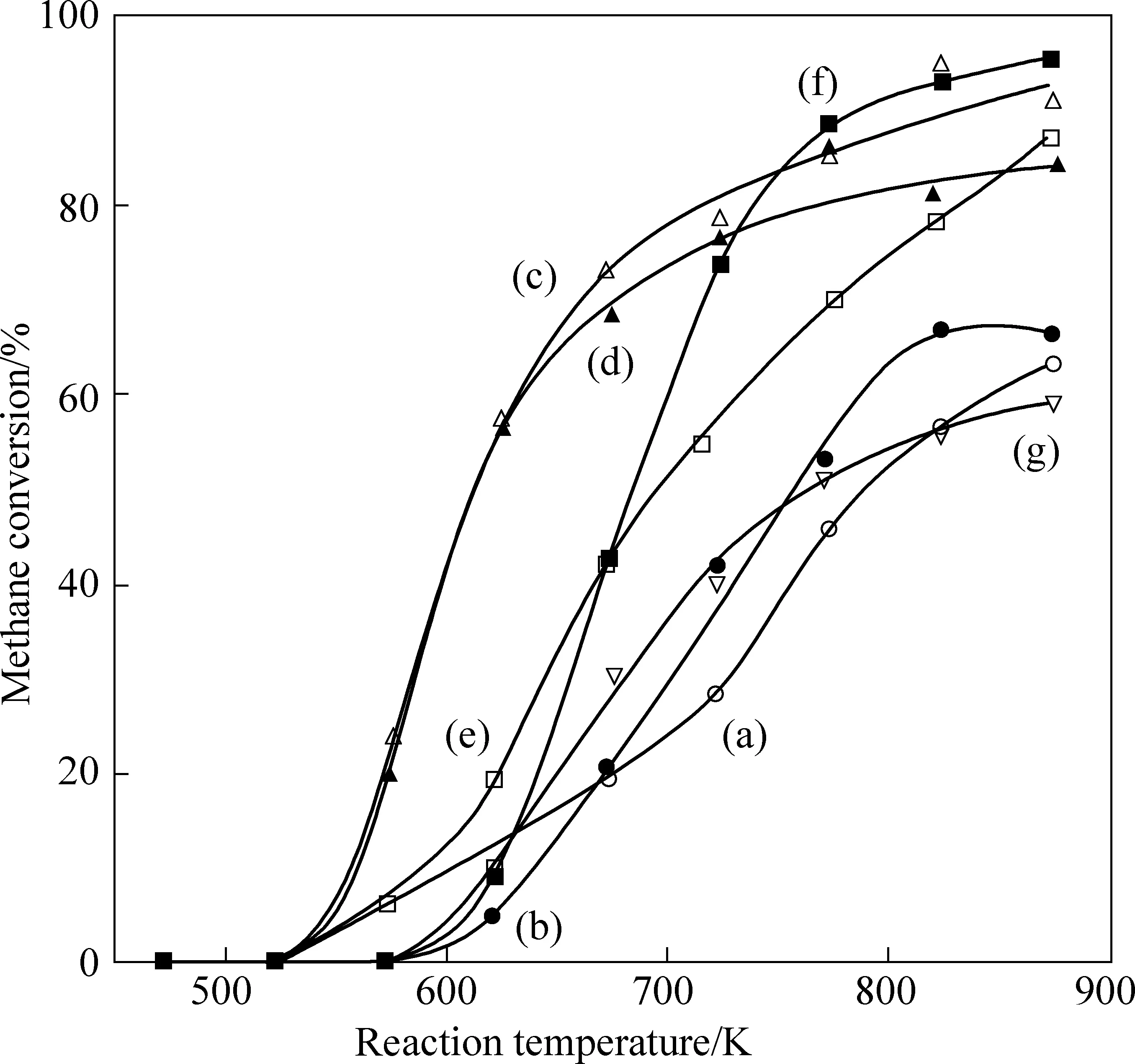
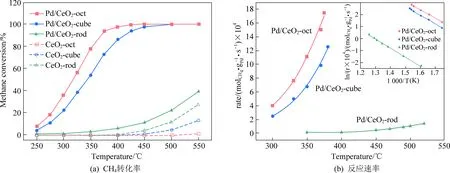
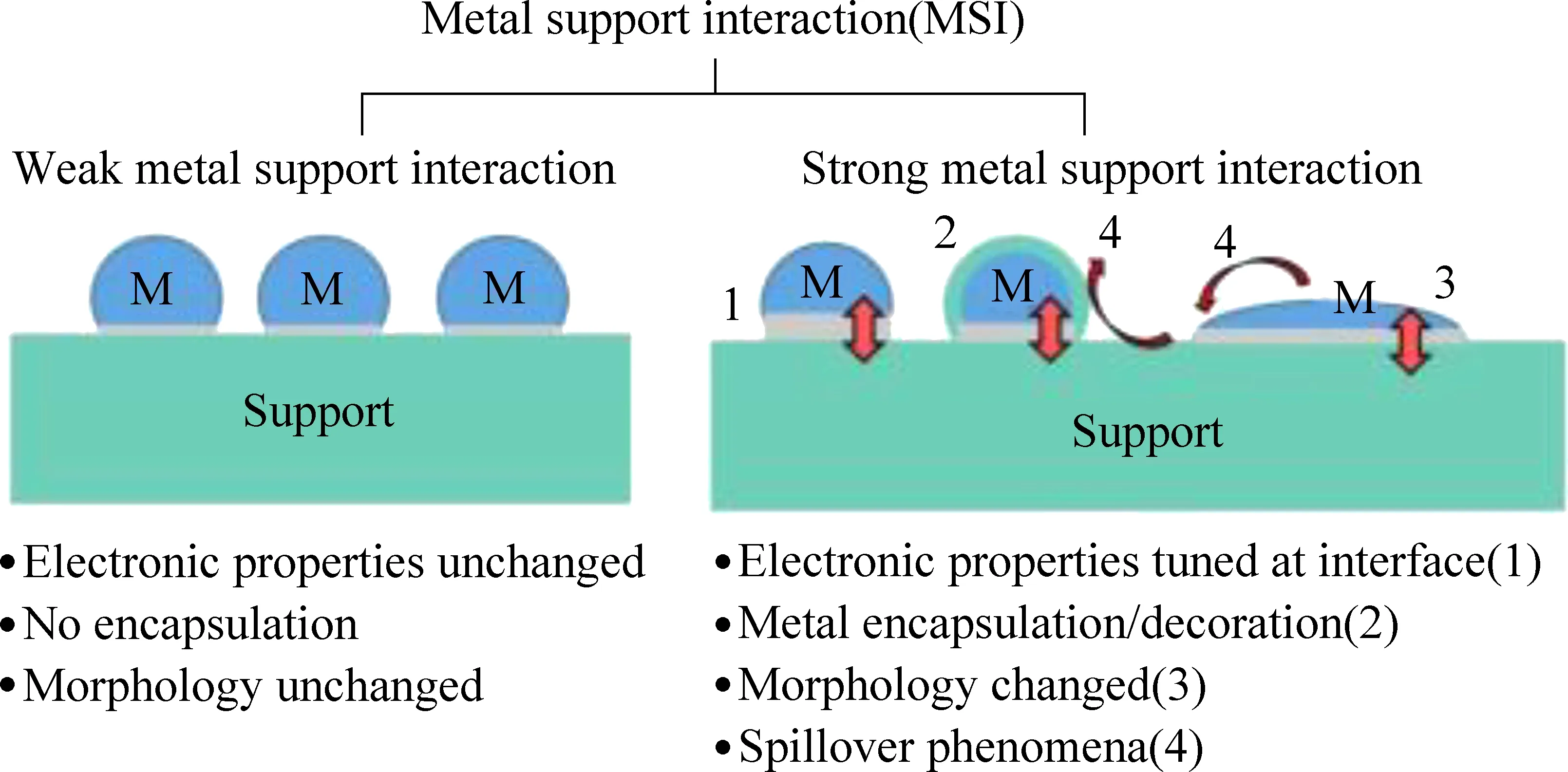
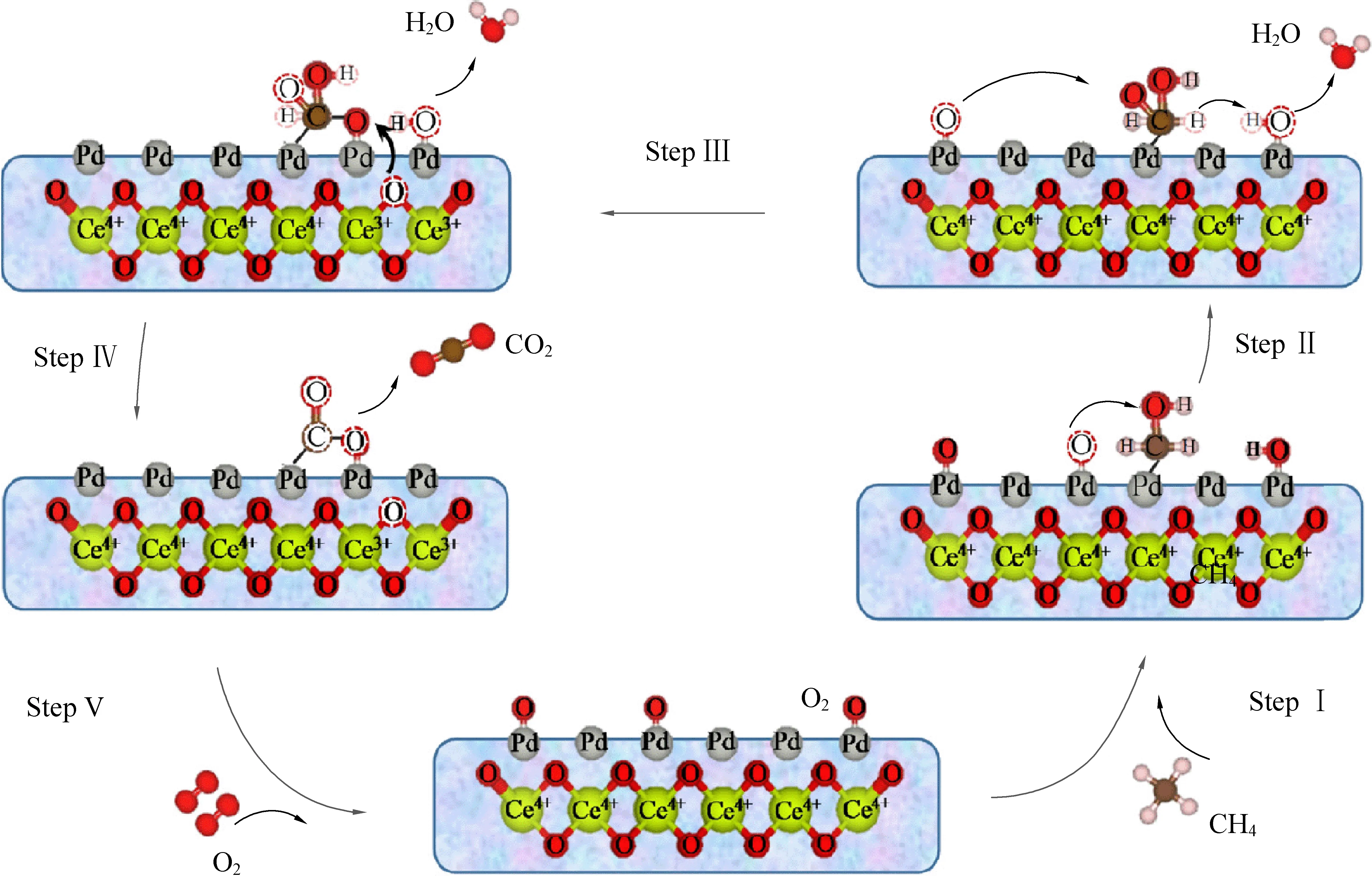
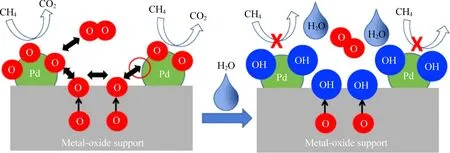
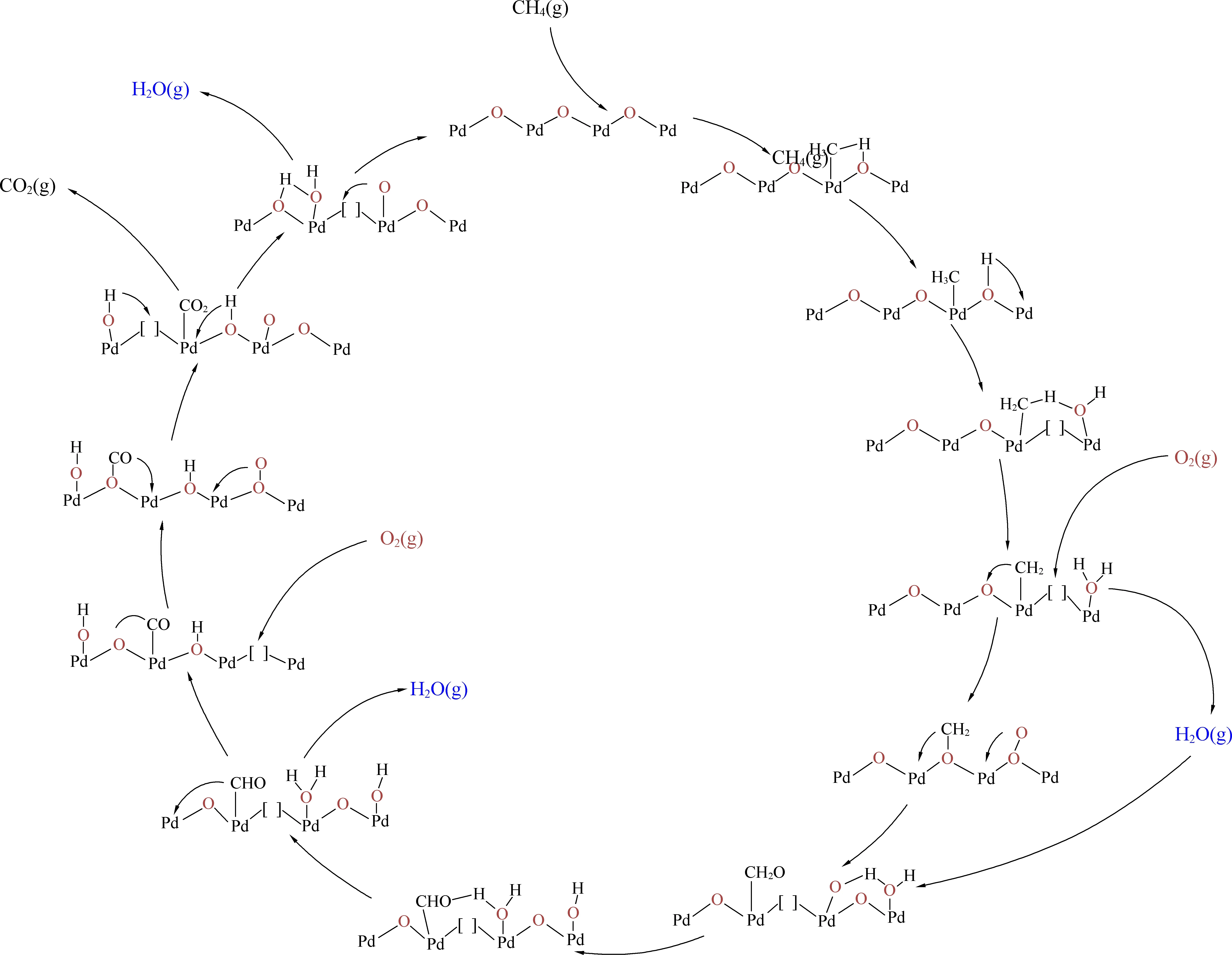
除了粒径之外,Pd在催化剂表面的分散态及价态分布也与反应活性密切相关。按元素价态划分,Pd在催化剂表面通常可以被分为:零价单质钯(Pd0)、高价氧化钯(PdO)以及非化学计量的钯氧化物(PdOx)(0 Huang等[5]采用初湿浸渍法制备了Ce-Zr改性的La-Al2O3催化剂,发现稀燃条件下Pd和PdO均可作为CH4氧化的活性位点,而富燃条件下仅有PdO起主导作用。Murata等[39]指出,金属氧化物载体上形成Pd@PdO核壳结构对CH4的活化效果最佳,说明Pd和PdO共同构成CH4氧化的活性位点。Xiong等[3]发现在过量的氧气中,经过还原后有Pd0存在的催化剂相比于氧化后的活性更高,并通过掺杂Pt使更多的Pd保持在还原状态,进而提升CH4氧化性能。 除了活性组分以外,载体性质对于CH4氧化过程也会产生影响,比如不同酸碱性的载体表面Pd位点的反应活性差异显著。Yoshida等[44]的研究表明,中等酸性氧化物(如ZrO2和Al2O3)上的Pd物种比强酸和碱性氧化物上的Pd物种反应性更强,如图1所示;Miller等[45]发现Pd/Al2O3的CH4反应活性优于Pd/CeO2和Pd/ZrO2-CeO2;而Eguchi等[46]证明Pd/SnO2的CH4氧化性能比 Pd/Al2O3、Pd/CeO2以及Pd/ZrO2-CeO2都要好。 (a) Pd/MgO, (b) Pd/ZrO2, (c) Pd/Al2O3 , (d) Pd/SiO2 , 图1 不同载体Pd基催化剂的CH4转化率[44]Fig. 1 Methane conversion over Pd-based catalystswith different supports[44] 然而,不同载体与Pd之间的相互作用强度会影响表面Pd颗粒大小,难以区分Pd颗粒结构和载体本身对于CH4氧化反应的贡献程度。为此,Willis等[20]将尺寸可控的Pd纳米粒子负载在不同载体表面,探究了载体效应对反应性能的影响。结果显示,以酸性(Al2O3)、惰性(SiO2)或可还原(CeO2-ZrO2)等金属氧化物作为载体时,催化剂CH4氧化速率接近;相比之下,碱性氧化物(MgO)载体的Pd基催化剂活性要低得多,推测Pd基催化剂载体对CH4氧化的作用可能与分子吸附对载体酸碱性质的依赖性有关(比如CO2中毒),也可能与Pd的电子状态有关。 此外,载体暴露的晶面也会影响CH4的氧化性能。Chen等[31]合成了纳米棒状、立方体以及八面体等不同形貌的CeO2载体并进一步负载Pd,研究了不同CeO2暴露晶面对于CH4氧化性能的影响,结果如图2所示。结果表明,对应于CeO2(111)晶面的八面体结构Pd/CeO2具备最高的CH4氧化活性,而对应于CeO2(111)和(110)晶面的棒状Pd/CeO2反应性能最低。不同形貌的CeO2载体上的Pd物种具备不同的分散状态:八面体上的Pd主要以二价PdO颗粒形式存在,立方体上的Pd主要以尺寸在1~2 nm左右的PdOx团簇形式存在,而CeO2棒上的Pd主要以高度分散的Pd4+离子形式存在。其中,Pd2+是CH4催化氧化的主要活性中心。Pd0的作用在于反应过程中可以逐渐氧化产生Pd2+,而CeO2晶格中的Pd4+对CH4氧化不发挥作用。 图2 不同CeO2载体的Pd基催化剂的和反应速率Fig. 2 CH4 conversion and reaction rate of Pd-based catalysts with different CeO2 supports 强金属-载体相互作用(Strong Metal-Support Interaction,SMSI)[12, 47-50]是催化剂设计方面的研究重点。与弱金属-载体相互作用(WMSI)相比,SMSI通常涉及表界面间的电荷转移、物种溢流、载体对金属的包覆以及组分形态变化等过程,会深刻影响Pd物种在载体表面的形态结构、暴露位点和分散状态,还可能会引起Pd团簇/颗粒被载体包覆或诱导Pd物种团聚,如图3所示[50]。基于SMSI效应对催化剂进行设计改性是调控催化剂活性、提高热稳定性以及改变活性位点数量的重要方式。 图3 弱金属载体相互作用和强金属载体相互作用之间的比较[50]Fig. 3 Comparison between weak metal supportinteraction and strong metal support interaction[50] 不同载体与Pd物种之间的SMSI效应强度不同,会起到抑制活性PdO热分解、减少活性组分烧结、提供活性氧物种等作用。比如,对于存在多种晶型结构的Al2O3,研究表明γ-Al2O3(100)晶面上的不饱和五配位Al3+位能锚定Pd物种,形成PdⅡ—O—Al键,改善Pd物种分散性。然而,α-Al2O3/θ-Al2O3表面的不饱和五配位Al3+较少,导致与Pd物种相互作用较弱[51-52]。Murata等[29]发现Pd与α-Al2O3或θ-Al2O3之间的弱相互作用使Pd颗粒具有较高比例的台阶位;而Pd与γ-Al2O3之间的SMSI效应则会阻碍球形Pd颗粒形成,最终影响催化剂的CH4氧化活性,如图4所示。 图4 不同晶型Al2O3上不同粒径Pd存在形式[29]Fig. 4 Existence forms of Pd with different sizes on different crystal forms of Al2O3[29] CeO2的储氧能力依赖于Ce4+←→Ce3+之间的快速切换,载体的氧化还原过程会影响Pd2+与Pd0的转化和Pd的烧结过程;反过来Pd物种也能对载体CeO2表界面产生不同的修饰作用,增加表面Ce3+或氧空位含量,有效提升催化剂的抗老化性能。研究表明,Pd物种可以作为单原子锚定在CeO2纳米棒(111)晶面;两者还可以形成Pd@CeO2核壳结构,利用SMSI效应提高界面晶格氧迁移率,具有较高的反应活性和热稳定性[53]。Colussi等[54]利用溶液燃烧法合成Ce1-xPdxO2-δ固溶体,发现形成了具有较高CH4氧化活性的相互作用相Pd-O-Ce有序表面结构,并将该结构的形成归因于SMSI效应下CeO2(110)复合重构,并且暴露出大量高活性氧空位。另外,载体的晶面也会影响Pd物种的价态分布。有研究发现Pd可以在CeO2表面上以PdO纳米颗粒的形式呈现Pd2+,也可以插入CeO2晶格中以Pdδ+(2<δ≤4)的形式存在[55-57]。 关于Pd基催化剂的CH4氧化反应机理,最早的认知建立在Eley-Rideal和Langmuir-Hinshelwoo两种机理模型上。根据Eley-Rideal机制,CH4或O2首先吸附在催化剂表面,之后与气相中的另一气体反应;而Langmuir-Hinshelwoo机制则描述了O2与CH4同时吸附在催化剂表面,然后表面活性氧物种与吸附态CH4之间反应的过程。随着表征技术的发展,研究者利用氧同位素标记法发现了催化剂表面晶格氧也会参与CH4的氧化过程,因而提出:催化剂表面Pd物种首先遵循Langmuir-Hinshelwoo机制将CH4和O2吸附在活性位点,然后通过Mars-van Krevelen机制催化CH4氧化[58],也是目前被普遍接受的反应机理。该过程主要是CH4首先吸附在表面Pd活性位点,位点上的晶格氧迁移形成表面活性氧与吸附的CH4反应,在释放产物后位点上会产生相应的氧空位,这些空位后续再由气相中的O2补充,完成整个催化循环,如图5所示。 图5 Pd-Ce-Zr催化剂上遵循Mars-van Krevelen机制的甲烷氧化过程[58]Fig. 5 Methane oxidation on the Pd-Ce-Zr catalyst following Mars-van Krevelen mechanism[58] 催化活性通常与反应速率决定步骤(RDS)有关。众所周知,CH4具有稳定的四面体结构,吸附在催化剂表面的CH4分子的第一个C—H键断裂通常被认为是速率决定步骤。之后会发生连续脱氢过程,伴随着甲基(CH3—)和亚甲基(—CH2—)生成,并与活性氧结合生成甲酸盐或碳酸盐等中间产物,最终生成CO2。Stotz等[59]利用密度泛函理论(DFT)研究了CH4在PdO(101)晶面上的反应机理,判断出表面存在Pd(cus)和O(cus)两个不饱和活性位点,并明确了在200~400 ℃温度范围内干燥条件下详细的C1反应路径,如图6所示,具体过程为:CH4(g) → CH3-Pd →CH2OH-Pd→CH2O-Pd→CHO-Pd→CO-Pd→CO-O→CO2(g)。 图6 200~400 ℃干燥条件下PdO(101)上甲烷氧化优先反应路径的催化循环[59]Fig. 6 Catalytic cycle of the preferential reaction path for methane oxidation over PdO(101) as found at dry feed conditions (200~400 ℃)[59] 虽然利用Pd与CeO2载体间的SMSI效应实现Pd—O—Ce的键合可以有效抑制Pd物种的烧结团聚,但这种SMSI效应也可能会引发催化剂的失活。与SMSI效应有关的失活方式可能是电子效应[50],例如Pd物种电子云密度改变而引发的价态变化;也可能是几何效应,例如Pd物种活性表面被载体包覆,尤其是当催化剂在高温下长时间运行,同时排气中存在H2、HC等还原性气体时更容易发生,如图7所示[60]。另外,高温下长时间运行后,载体中的Al2O3可能会发生相变,由高活性的γ相转变为低活性的α相,这也是催化剂失活的主要原因之一。 图7 Pd物种对Ce0.5Zr0.5O2表界面修饰提升催化剂抗老化性能[60]Fig. 7 Pd species modified Ce0.5Zr0.5O2 surface to enhance anti-aging properties of catalysts[60] 1971年,Cullis等[61]发现Pd/Al2O3催化剂的CH4反应速率随水蒸气浓度的增加而下降,并提出可能形成了表面羟基从而对活性位点产生抑制作用的观点。后续研究表明,在450 ℃以下,H2O比CH4更容易吸附在Pd活性位点上,H2O的解吸成为CH4氧化的速率控制步骤,并且H2O可能会导致Pd物种形成Pd-OH后发生迁移团聚[21, 62-63]。Ciuparu等[64]利用同位素标记法发现潮湿氛围PdO/ZrO2上的表面活性氧与气相氧之间的同位素交换受到阻碍,而当空位被H2O堵塞时,气相氧的吸附可能受到表面吸附水分子的限制。 Ciuparu[65]的另一项研究发现表面Pd(OH)2的积累会干扰表面活性氧的迁移过程,如图8所示。 Li等[66]利用常压X射线光电子能谱研究了潮湿氛围下催化剂表面Pd0、PdOx和PdO的演化过程,发现Pd-OH占据了CH4吸附的不饱和位点以及CH4活化的相邻氧化位,并且甲基反应中间体的脱氢过程也受到抑制,最终限制了CH4的氧化活性。Velin等[67]采用原位漫反射傅里叶红外光谱研究了PdO/Al2O3表面羟基的形成与CH4氧化活性之间的关系,发现了表面羟基化遵循两种不同的途径:1)含氢物质溢流到PdO/Al2O3边界;2)含氢物质溢流到PdO颗粒附近。其中,第二条路径是低温下催化剂水中毒的主要原因。 图8 Pd基催化剂的H2O中毒失活机制[66]Fig. 8 H2O poisoning deactivation mechanism ofPd-based catalyst[66] 此外,Stotz等[59]利用密度泛函理论提出了在300~550 ℃潮湿(体积分数12%H2O)氛围下CH4的氧化路径遵循以下过程:CH4(g) → CH3-Pd → CH2-Pd → CH2-O → CHO-Pd → CO-Pd → CO-O → CO2(g),如图9所示[68]。与无水蒸气时的反应路径相比,主要区别于高温下没有生成羟基甲基中间体而是直接产生了亚甲基。这可以归因于Pd(cus)位点被表面羟基覆盖的抑制作用。 图9 300~550 ℃潮湿条件下PdO(101)上甲烷氧化优反应路径[59]Fig. 9 The optimal reaction path of methane oxidation on PdO (101) under 300~550 ℃ humidity[59] 尽管天然气发动机排气中的硫含量很低(约1 ppm),催化剂长期暴露在含硫的废气中依然会中毒失活。已经证实在200 ℃以上稀燃条件下,废气中的SOx会导致催化剂载体或活性组分发生硫酸化[69],如图10所示。与水中毒类似,硫中毒后的Pd基催化剂表面的高活性Pd不饱和位点会被占据而形成更多的PdO—SOx。此外,SOx还可以通过阻止CeO2中的Ce3+/Ce4+切换来抑制表面Pd物种与载体之间的氧迁移[69]。但与水中毒不同的是,SOx引起的催化剂中毒是不可逆的。这由于硫酸盐物种难以完全分解,硫中毒的Pd/Al2O3催化剂即使在650 ℃下也无法完全再生。 图10 富氧条件Pd/Al2O3硫中毒过程及再生机理[69]Fig. 10 Spoisoning and regeneration mechanisms for Pd/Al2O3 in O2-rich conditions[69] 影响催化剂抗硫性能的关键因素是载体对SOx的吸附。例如,Al2O3表面存在更多的SOx吸附位点,更容易发生硫酸化,从而有效减少SOx对活性位点的攻击,因此相比于MgO、SiO2等其他载体,催化剂的抗硫性能更好。而TWC催化剂中的CeO2同样能使硫酸盐优先在CeO2上形成,对表面的Pd物种起到保护作用[70]。 船用天然气发动机排气中低浓度甲烷(CH4)的高效去除是对当今气候变化的重要措施,也是后处理技术领域的研究重点。Pd基金属氧化物是目前应用最广泛的CH4氧化催化剂。鉴于发动机排气温度低、CH4化学稳定性高、尾气环境恶劣以及催化剂贵金属用量大等问题,低成本设计开发更多低温高效、抗水抗硫性能优异、耐久性高的催化剂是未来的重点研发方向。因此,需要继续从分子层面对催化剂结构进行设计。活性组分Pd的分散状态、暴露晶面、粒径尺寸、价态分布、PdO与金属Pd之间的相变、载体种类、金属-载体间相互作用以及组分间的协同效应均会影响催化剂的甲烷氧化性能,需要充分平衡不同因素对反应性能的影响利弊,建立催化剂结构与性能之间的构效关系,在明晰Pd基催化剂甲烷氧化机理以及失活机制的基础上可以采用表面活性位点锚定、引入第二活性位点、添加助剂以及适当的封装包覆等多种方式,构筑高效稳定的表面活性位点,以获得低温活性和可靠性兼备的高性能甲烷氧化催化剂。3.3 载体对催化活性的影响
3.4 强金属-载体间相互作用对催化活性的影响

4 Pd基催化剂的甲烷氧化机理研究

5 Pd基催化剂的失活机制研究
5.1 高温失活机制

5.2 水中毒机制
5.3 硫中毒机制

6 前景展望
