光皮树SNP高密度遗传图谱构建
2023-05-04吉悦娜张路红陈韵竹李培旺李党训陈景震
吉悦娜,夏 栗,曾 霞,张路红,3,陈韵竹,杨 艳,李培旺,李党训,陈景震
(1.湖南省林业科学院省部共建木本油料资源利用国家重点实验室,湖南 长沙 410004; 2.湖南财政经济学院,湖南 长沙 410205; 3.中南林业科技大学生命科学与技术学院,湖南 长沙 410004)
木本油料植物泛指富含天然油脂的灌乔木,随着经济与科学技术的发展,植物油脂用途不断拓展,植物油脂市场需要逐步壮大,在国民经济中起着不可替代的作用[1]。筛选、培育高产、高含油、适性强的木本油料植物资源成为当前的研究热点之一。光皮树(Swidawilsoniana)为山茱萸科(Cornaceae)梾木属(Swida)落叶乔木[2]。该树种抗逆性、生态适应性强,果实含油率较高,且加工性能好,不仅是优质的木本食用油树种,亦是品质较高的多用途工业原料油树种,其嫁接苗3 a后开始挂果,结果时间长、产量高,丰产期油脂产量可达1 125~1 500 kg·hm-2[3-6]。目前光皮树优良品种的选育主要采用传统的自然选种、杂交育种以及倍性育种等手段,个体间树体生长势、开花结果特性、果实含油率等性状均存在较明显差异[7]。由于选育周期长,且缺乏遗传研究基础,对高产、高含油、高抗等表型优异的功能基因的挖掘筛选不足,限制了其优良新种质的开发及其推广应用。
简化基因组测序(reduced representation genome sequencing,RRGS),是利用限制性内切酶打断基因组DNA,对特定片段进行高通量测序获得海量遗传多态性标签序列来充分代表目标物种全基因组信息的测序策略。其实验方法步骤简单,成本低,而且可以不依赖参考基因组就能获得全基因组范围内的遗传多态性标签,因而广泛应用于生态学、进化学和基因组学等领域[8]。单核苷酸多态性标记(simple nucleotide polymorphism,SNP)为第三代分子标记最早由 Lander 提出,因其高效、快速、稳定、可靠等诸多优点,在物种遗传多样性和进化分析、高密度遗传图谱的构建以及数量性状位点(quantitative trait loci,QTL)定位、基因组关联分析等研究领域得到广泛应用[9]。在光皮树上关于遗传图谱构建的研究起步较晚,前期分子水平研究主要集中在ISSR、SSR等分子标记开发上面[10-12],但存在上图量少、标记间遗传距离较大、基因组覆盖率低等缺点,而SNP标记的上图量远远超过传统分子标记,图谱分辨率高、定位精度高[13]。例如,Sun等[14]通过全基因组重测序构建了杨树(Populus)的高密度遗传图谱,得出该连锁图谱由19个连锁群(LGs)的5 796个SNP标记组成,平均标记距离为0.46 cM,确定了数量性状位点(QTL),其研究结果有助于植物育种家培育高生物量基因型杨树。徐礼羿[15]研究了油茶(Camelliaoleifera)遗传图谱,开发了适用于 327 个茶树作图群体基因分型的 SNP 标记。King等[16]应用来自美洲、非洲和东南亚的多个麻疯树(Jatrophacurcas)种质进行种内杂交,构建了4个F2作图群体,随后SNP标记构建了连锁图谱,有助于麻疯树农艺性状的QTL定位及分子辅助育种研究。
近年来,随着几代测序技术、分子标记技术的发展和一些针对林木作图策略的提出,林木的遗传图谱构建研究进展迅速。本研究基于课题组前期对光皮树种质资源的大量研究,选取农艺性状差异较大的光皮树亲本及其F1代为原材料,基于全基因组重测序技术和生物信息学分析方法对它们进行SNP标记多态性分析,构建光皮树高密度遗传图谱,旨在为光皮树多种农艺性状的QTL定位和基因挖掘奠定良好的基础。
1 材料与方法
1.1 试验材料
试验材料来源于湖南省林业科学院实验林场光皮树种质资源收集圃,为光皮树优良无性系父本XL-G4和母本XL-G1亲本及其杂交的121株F1代幼苗。采集健康幼嫩的叶片,放入液氮罐速冻带回实验室,于-60 ℃保存待用。
1.2 DNA提取
采用新型DNA 提取试剂盒(天根生化科技(北京)有限公司,试剂盒型号DP320-02)提取DNA,严格按照其使用说明步骤操作。DNA纯度检测采用NanoDrop方法,其OD260/OD280范围在 1.7~1.9 视为合格。DNA完整性检测用1%琼脂糖凝胶电泳,1×TAE缓冲液,电压5 V·cm-1,电泳时间30 min。
1.3 简化基因组测序与建库
光皮树DNA测序工作委托上海美吉生物科技医药有限公司完成。采用RAD建库方式构建pair-end文库,长度范围在300~500 bp,用Illumina HiSeq PE50进行测序。
1.4 SNP分子标记的开发
首先,将样本的RAD-tag进行比较、对照和归类,按照不同tag的深度信息对其进行排序(降序);随后将各个样本内部的RAD-tag进行比对获取其杂合位点信息,进一步将不同样本之间的RAD-tag进行比对搜索个体之间的单碱基差异信息;最后,通过各个样本的RAD-tag频数表和比对表信息,删除可能来自重复区域的结果,得到高可信度的群体SNP标记基因分型结果,标记类型为hk×hk、lm×ll、nn× np,即亲本一方纯合,一方杂合,或均杂合,并最后对其校正后的数据进行再次过滤与筛选。
1.5 高精度遗传图谱构建
通过SNP标记基因分型结果与统计将所得的标记用于构建光皮树SNP遗传连锁图谱。使用软件Joinmap 4.0对过滤筛选获得的标记进行聚类分析,采用LOD=8进行分群构建父母本的连锁图谱,获得连锁群。
1.6 数据分析软件
利用SPSS Statistics 17.0中文软件进行分析。
2 结果与分析
2.1 光皮树杂交F1代幼苗DNA质量检测
对测序样品的DNA质量进行把控是简化基因组测序的前提条件,高质量的光皮树DNA为后续的数据分析提供可靠的基础。使用NanoDrop进行光皮树DNA纯度检测,Qubit对其浓度进行检测。所提取的样本DNA,OD260/OD280大于1.60,浓度大于10 ng·μL-1,且样品量大于500 ng,满足简化基因组文库建库需求。
2.2 简化基因组测序原始数据质控
运用统计学的方法,采用软件Trimmomatic(http://www.usadellab.org cmsuploads/supplementary/Trimmomatic)对所有测序reads的每个circle进行碱基分布和质量波动的统计,可以直观地反映出样本的测序质量和文库构建质量。图1为原始数据的碱基组成分布图,序列的前后位置与测序的引物接头相连,碱基A、T、G、C在起始端会有所波动,后面趋于稳定。该文库碱基A与碱基T数量趋于平衡,碱基G与碱基C也相近,说明各碱基在此文库分布均匀。
原始数据reads的碱基质量是后续试验的基础和保障,碱基质量越高说明测序过程中模糊碱基(unkown bases,N)就越少,reads错误率越低。碱基的排列(5’-3’端)在0~95之间碱基质量值基本上都大于36;碱基的排列(5’-3’端)在95~120之间碱基质量值有所下降;碱基排列在125以后又趋于稳定,碱基质量值在32左右(见图2)。总体来看,所有reads碱基的综合质量较高,为后续试验的准确性提供了保障。
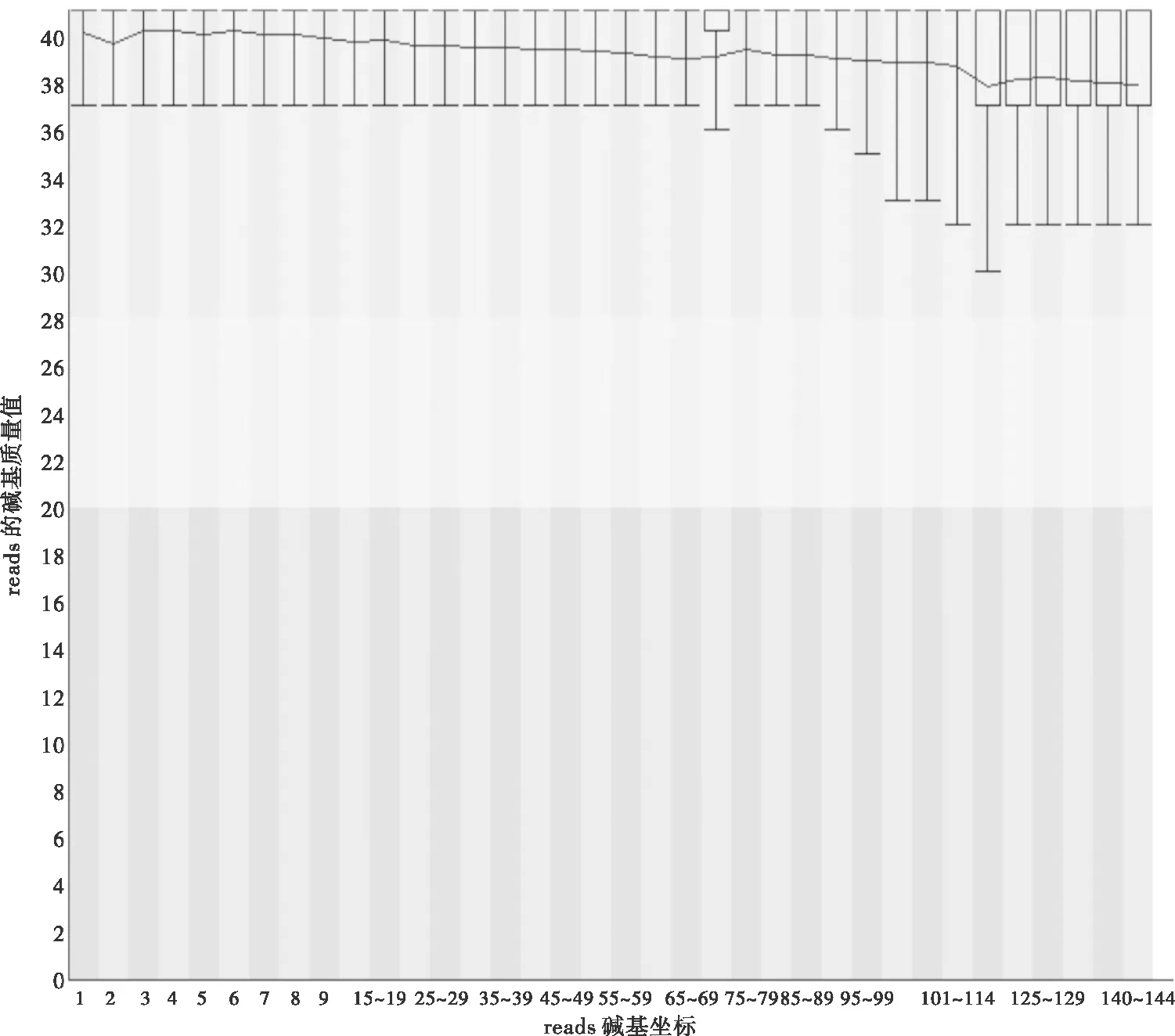
图2 碱基质量分布图(PE150)Fig.2 Bases mass distribution (PE150)
2.3 简化基因组测序原始数据统计
对光皮树进行conting denovo组装。为了保证数据分析的可靠性与准确性,需要对测得的原始数据进行筛查和过滤,以此来获取到高质量的clean data以保证后续生信分析的顺利开展。通过软件进行数据过滤统计,各个样本总的序列和碱基数据均比较大。碱基GC的所占总数的百分比为:36.22% ~ 37.2%;Q20最大值为98.40%,最小值为97.84%;Q30最大值为95.16%,最小值为93.80%。即过滤筛选后,碱基错误率大大降低,质量大大得到提高,保证了生物信息分析的准确性。
2.4 RAD-tag文库构建与深度统计
为使接下来的SNP分析更具可靠性,做RAD-tag分析时,应遵循以下过滤条件对数据进行筛选和过滤,即RAD-tag序列中不允许存在模糊碱基(N),起始端(5’)非酶切位点(AATTC)起始的序列。建立121个子代单株RAD文库,子代单株RAD文库中含有1 177 021 204条原始序列,父本XL-G4含有14 808 894条原始reads,母本XL-G1含有14 219 880条原始reads,PE150(5’-3’端)的reads中在105 bp以后序列碱基的测序质量出现较为明显波动,去除reads中105 bp以后的读段。随后根据相同酶切处RAD-tag测序reads之间的序列全同性(stacks软件,ustacks程序),将reads进行统计并获得各个样本RAD-tag的数目和深度信息,用于评估RAD技术对于酶切区域的富集效果。最终原始序列经过质量控制过滤,子代单株RAD文库保留1060573106条高质量reads,高质量reads占有比率90.11%;父本XL-G4有13277760条高质量reads,母本XL-G1保留12780718条高质量reads,父母本保留高质量序列比例分别达到89.66%和89.88%;最终RAD-tag文库中高质量序列达到了1 086 631 584条,比率达到了90.10%,满足测序文库中高质量序列达到原始序列80%以上的要求,满足了RAD文库的构建要求(见表1)。

表1 RAD-tag文库测序结果统计Tab.1 Sequencing results of rad tag library样品原始序列高质量序列Q20/%Q30/%比率/%父本14 808 89413 277 76095.5890.7189.66母本14 219 88012 780 71895.5490.6389.88F1子代1 177 021 2041 060 573 10694.7989.8990.11合计1 206 049 9781 086 631 58490.10
对光皮树无性系杂交的2个亲本和121个F1子代进行RAD-tag聚类并统计RAD-tag 数据显示,深度值RAD-Tag、Depth>1、Depth>4,Depth>9的最大频数值依次为:2133541、740494、528903、341909,最小频数值分别为:468570、186751、27824、2925,平均频数值为:983331、465442、212989、65226,表现为深度值越大,频数值越小(见图3)。
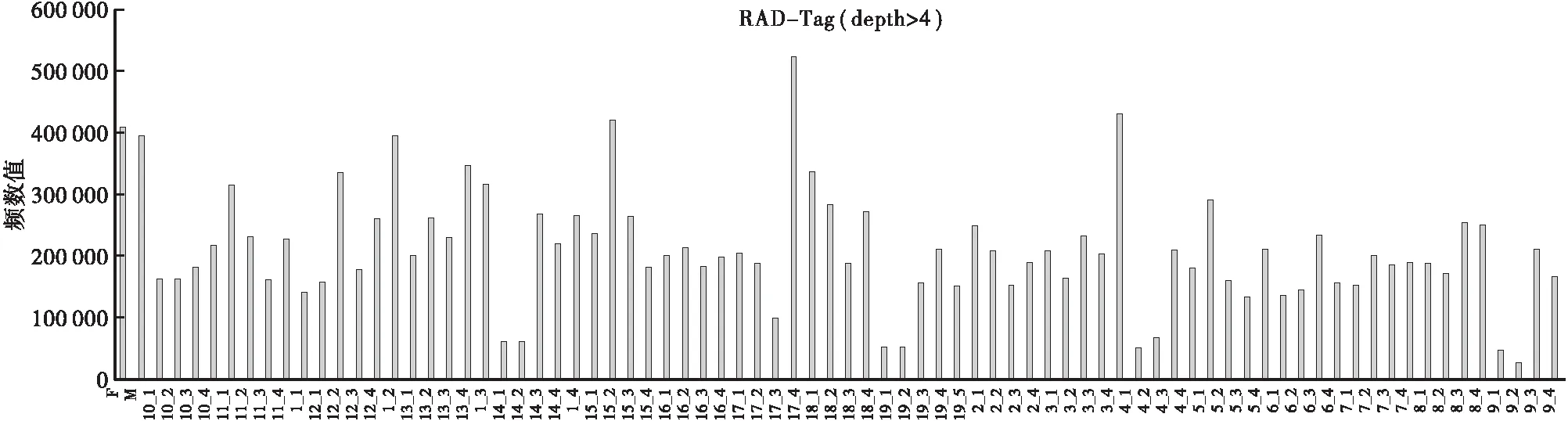
图3 Depth>4频数统计图Fig.3 The frequency statistics of Depth>4
2.5 SNP标记基因分型结果与统计
在未进行校正前得到基因型分类结果为hk×hk、nn× np、lm×ll的数量分别是:8707个、23095个、25287个,共57089个。校正后基因型hk×hk、nn× np、lm×ll数量分别是:8707个、23087个、25277个,此时共有57071个标记,hk×hk标记无变化,nn× np删减标记8个(nn× np 97、nn× np 285、nn× np 341、nn× np 450、nn× np 575、nn× np 936、nn× np 4336、nn× np 4856),lm×ll删减标记10个(lm×ll 207、lm×ll 266、lm×ll 498、lm×ll 1 245、lm×ll 1427、lm×ll 1578、lm×ll 1992、lm×ll 2083、lm×ll 2705、lm×ll 4112)(见表2)。

表2 SNP标记基因分型与校正结果Tab.2 SNP marker genotyping and correction results基因型校正前数量校正后数量校正后结果(删除标记)hk×hk8 7078 707无删除标记lm×ll25 28725 277lm×ll 207、lm×ll 266、lm×ll 498、lm×ll 1 245、lm×ll 1 427、lm×ll 1 578、lm×ll 1 992、lm×ll 2 083、lm×ll 2 705、lm×ll 4 112nn×np23 09523 087nn×np 97、nn×np 285、nn×np 341、nn×np 450、nn×np 575、nn×np 936、nn×np 4 336、nn×np 4 856合计57 08957 07118
为获得可信度高的遗传图谱可用标记,需要对其校正后的数据进行再次过滤与筛选。过滤与筛选后共剩余标记9110个,占过滤前的15.96%。hk×hk、nn×np、lm×ll基因型分别为2490个、3353个、3267个,分别占过滤前的28.59%、14.52%、12.92%。
以过滤筛选后的9110个标记进行分型,将121个F1子代与筛选过滤后的SNP标记tag进行比对,通过统计F1子代SNP基因型(hk×hk、nn×np、lm×ll),得到F1子代SNP标记基因分型结果。2490个hk×hk基因型中hh、hk、kk分型数分别为39942个、79814个、39866个,分别占此基因型总数的21.08%、42.14%、21.05%。3353个nn×np基因型中nn、np分型数分别为101124个、105619个,分别占此基因型总数的40.73%、42.54%。3353个基因型为lm×ll中ll、lm分型数分别为111453个、101382个,分别占此基因型总数的43.69%、39.74%。分型hh、hk、kk、nn、np、ll、lm、未分型,所占总分型的百分比分别为:5.77%、11.52%、5.75%、14.60%、15.25%、16.09%、14.63%、16.40%。
2.6 光皮树遗传图谱构建
两个亲本连锁群内SNP标记数为579~1744个,平均连锁群 991个SNP标记,一共有SNP标记数量为8921个(见表3)。其中父本XL-G4 9个连锁群遗传距离从74.90到273.75 cM不等,覆盖图距为1277.55 cM;母本XL-G1 9个连锁群遗传距离从48.25到198.55 cM不等,覆盖图距为1011.47 cM;父母本SNP标记个数都是一样的,都是8921个,但是父本XL-G4平均标记距离为0.14 cM,母本XL-G1平均标记距离为0.11 cM,表明母本XL-G1连锁群体标记密度明显高于父本XL-G4连锁群。光皮树父本、母本SNP标记连锁图见图4和图5。
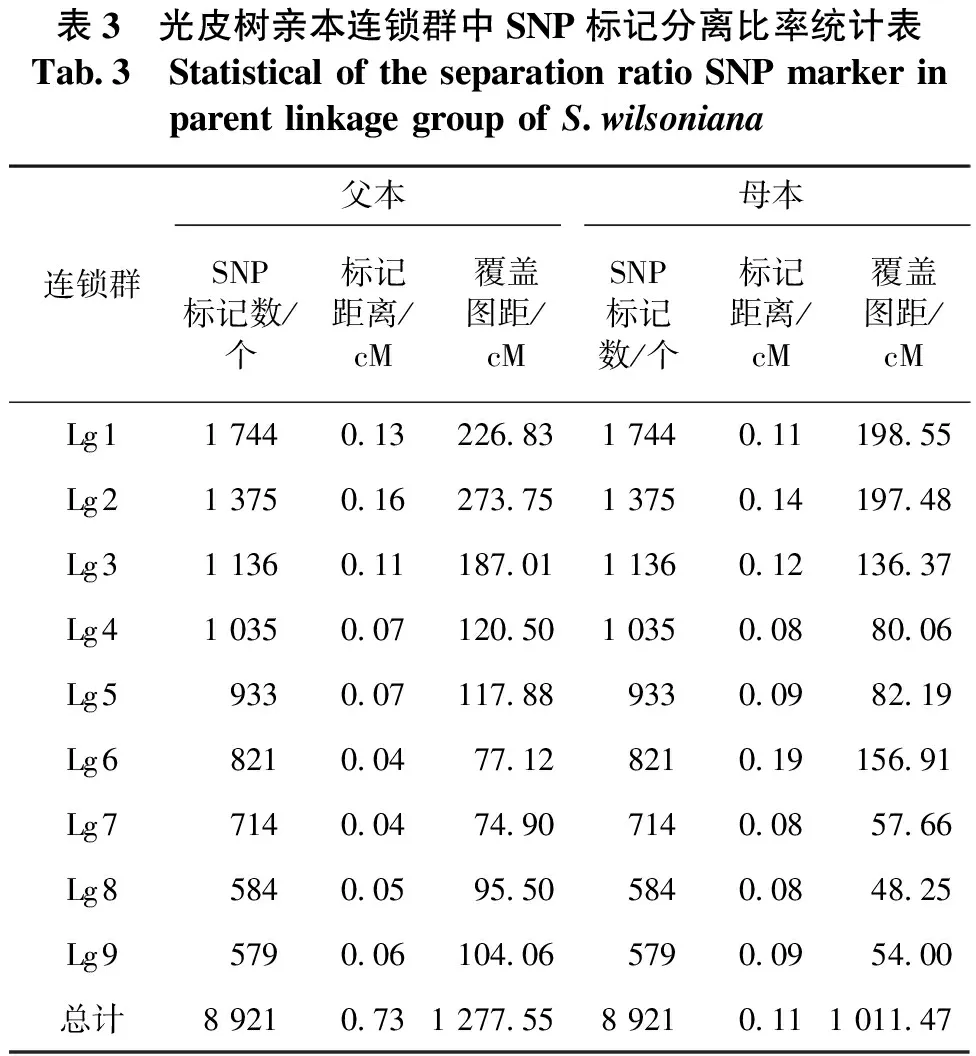
表3 光皮树亲本连锁群中SNP标记分离比率统计表Tab.3 Statistical of the separation ratio SNP marker in parent linkage group of S.wilsoniana连锁群父本母本SNP标记数/个标记距离/cM覆盖图距/cMSNP标记数/个标记距离/cM覆盖图距/cMLg11 7440.13226.831 7440.11198.55Lg21 3750.16273.751 3750.14197.48Lg31 1360.11187.011 1360.12136.37Lg41 0350.07120.501 0350.0880.06Lg59330.07117.889330.0982.19Lg68210.0477.128210.19156.91Lg77140.0474.907140.0857.66Lg85840.0595.505840.0848.25Lg95790.06104.065790.0954.00总计8 9210.731 277.558 9210.111 011.47

图4 光皮树父本 SNP 标记连锁图Fig.4 The S.wilsonianamale parent SNP marker linkage map
将两个亲本的分离比率进行整合,进行综合遗传图谱统计(见表4)。9个连锁群综合遗传距离从93.26到241.02 cM不等,综合覆盖图距为1 260.94 cM,平均每个连锁群1 091个标记;标记数最大的连锁群是Lg1,标记数为1 744个,标记数最小的连锁群是Lg9,只有579个;9个连锁群标记距离0.11~0.18 cM不等,平均标记距离0.14cM,标记距离最大和最小的连锁群分别是Lg 2和Lg 4,标记距离分别为0.18 cM和0.11cM。通过SNP 标记数、标记距离和覆盖图距3个参数指标进行相关性分析,SNP 标记数与覆盖图距呈极显著正相关,相关系数0.907,其中SNP 标记数超过1 000个标记的连锁群包括Lg 1、 Lg 2、Lg 3和Lg 4,覆盖图距均超过了110 cM。通过整合后的数据构建遗传图谱连锁群图(见图6)。
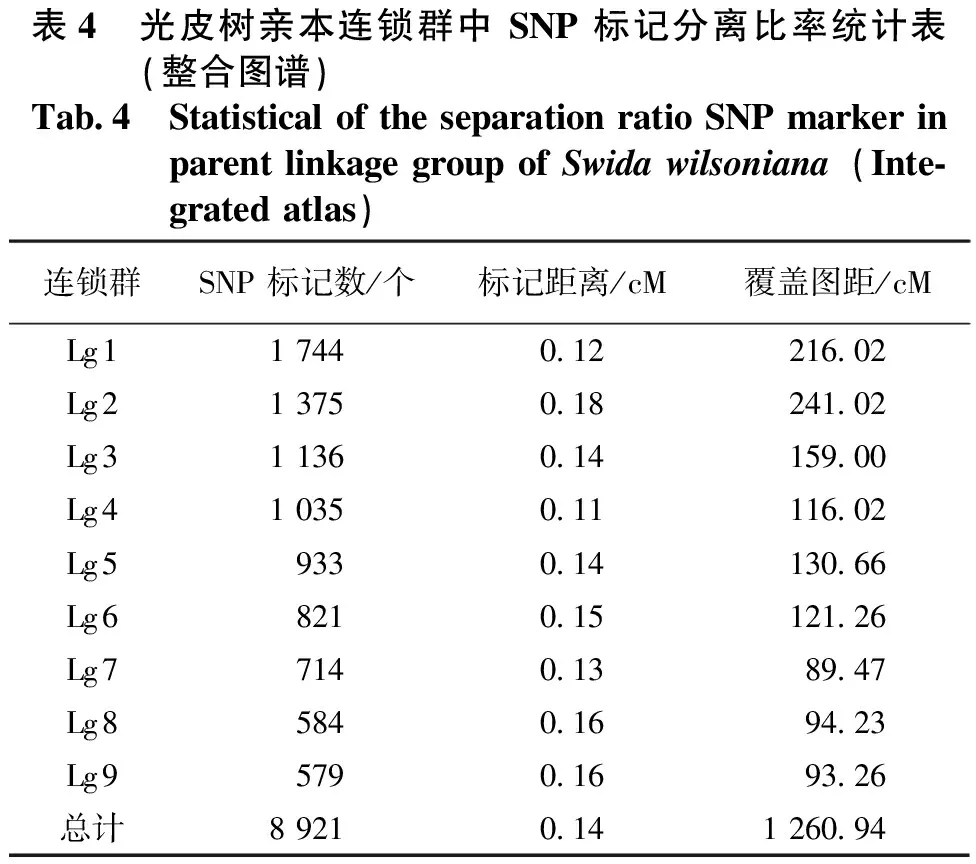
表4 光皮树亲本连锁群中 SNP 标记分离比率统计表(整合图谱)Tab.4 Statistical of the separation ratio SNP marker in parent linkage group of Swida wilsoniana(Inte-grated atlas)连锁群SNP 标记数/个标记距离/cM覆盖图距/cMLg11 7440.12216.02Lg21 3750.18241.02Lg31 1360.14159.00Lg41 0350.11116.02Lg59330.14130.66Lg68210.15121.26Lg77140.1389.47Lg85840.1694.23Lg95790.1693.26总计8 9210.141 260.94
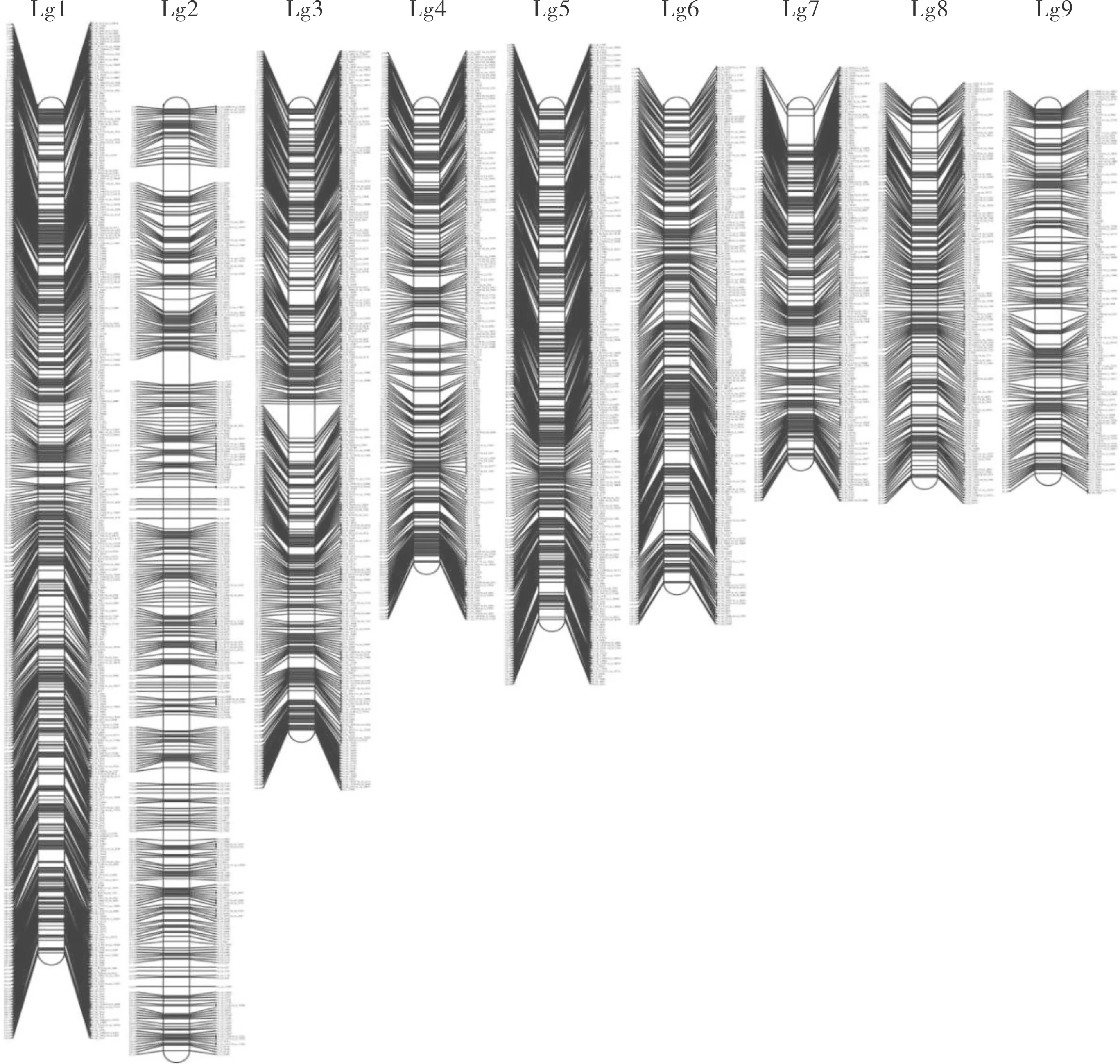
图6 光皮树亲本 SNP 标记连锁图(整合图谱)Fig.6 The Swida wilsoniana parental SNP marker linkage map(Integrated atlas)
3 结论与讨论
遗传连锁图谱是研究植物性状基因定位的重要组成部分,其中作图群体的构建尤为关键。而基于高密度遗传连锁图谱的QTL定位,是数量性状遗传研究的重要方法,标记和QTL是连锁的,也是分子标记辅助育种和目的基因分离与克隆的重要前提,对缩短育种周期、有目的性的选育新品种材料等具有重要意义。近年来,遗传图谱构建和QTL定位研究为不少国内外育种专家关注的重点。在非油料植物的连锁遗传图谱方面,乔东亚等[17]利用SNP标记对85份紫薇(Lagerstroemiaindica)种质资源进行聚类分析,探究其遗传多样性,筛选得到了高质量的SNP标记,并进一步通过对SNP分型数据进行遗传多样性分析。Tong等[18]利用 2 545个高质量的 SNP 标记分属美洲黑杨(Populusdeltoides)“1-69”和小叶杨(Populussimonii)“L3”两张高密度连锁图,分别为 20 个连锁群。唐海霞等[19]利用已构建的 103 株“冬枣”ד金丝 4 号”F1杂交群体和 145 株“冬枣”ד中宁圆枣”F1杂交群体,采用简化基因组测序技术,开发了SNP标记,构建了枣(Ziziphusjujuba)的高密度集成遗传连锁图谱。史晓畅等[20]以果实大小和果实硬度都存在显著性差异的山楂(Crataeguspinnatifida)品种“山东大绵球”(母本)和“新宾软籽”(父本)创建杂交群体,选用 130 株杂交 F1 代为作图群体,采用 RAD 测序技术开发的 SNP 标记构建山楂高密度分子遗传图谱。董明亮[21]以华北落叶松(Larixprincipis-rupprechtii)F1代为作图群体,通过SNP标记建立华北落叶松高密度遗传连锁图谱,依据连续两年测得的表型数据,将QTL定位应用于8个重要性状分析。而在油料植物的连锁遗传图谱相关研究中,高凤云等[22]基于亚麻(Linmusitatissimum)植物构建的单核苷酸多态性连锁遗传图谱,采用CIM对13个性状进行QTL定位,结论得出有20个与粗脂肪及其组成成分相关的QTL。滕卫丽等[23]利用ICIM法作加性数量性状座位和数量性状座位间上位性互作检测,获得81个与大豆(Glycinemax)粒形性状相关的QTL。李文杨等[24]利用筛选到的单核苷酸多态性SNP位点对作图群体进行基因型分析,构建了泡桐(Paulowniafortunei)连锁遗传图谱,此研究为泡桐分子育种学研究奠定了基础。
本课题研究在构建光皮树F1代群体基础上,通过多样性分析进行分子标记目标性状,目的是在早期目标性状定位之后,实现对木本油料光皮树的精准育种。本研究利用光皮树杂交F1代幼苗群体构建作图群体,首次定位标记出57 089个SNP,通过过滤与筛选后共剩余标记9 110个,基本实现了多态性分布和密度高的要求。进一步通过研究基本构建了光皮树遗传连锁图谱,对F1代幼苗表型性状进行遗传连锁标记,分布在9个连锁群中,遗传距离在93.26~241.02 cM之间,综合覆盖图距为1 260.94 cM,平均每个连锁群1091个标记,Lg1为标记数最大的连锁群(1 744个),Lg9为标记数最小的连锁群(579个)。这些 QTLs 簇的区域是后续挖掘光皮树相关性状功能基因需要重点关注的区域。本研究亦可为往后对光皮树良种选育、相近木本油料植物遗传图谱的构建策略制定提供参考。
