基于多层功能结构的谷物蛋白质功能预测
2023-03-31沈婷婷
沈婷婷,刘 静,管 骁
基于多层功能结构的谷物蛋白质功能预测
沈婷婷1,刘 静1※,管 骁2,3
(1. 上海海事大学信息工程学院,上海 201306; 2. 上海理工大学健康科学与工程学院,上海 200093;3. 国家粮食产业(城市粮油保障)技术创新中心,上海 200093)
为使研究人员可以更加便捷、准确地选择功能蛋白质,更高效完成谷物功能性食品的研发与创新,该研究提出基于多层功能结构的谷物蛋白质功能预测方法。该研究首先构建多种谷物数据共建的大规模相互作用网络,通过集群的功能特征与未知蛋白的交互作用探寻未知蛋白的相关功能;其次,定义新的蛋白质权重与语义相似度、功能层级权重来确定蛋白质可能具有的功能;最后,通过评分机制辅助完成谷物蛋白质功能的预测结果的判定。试验结果表明,该研究提出的预测方法使预测的功能具有层级性的特点,并且可获得指定功能蛋白质;对功能类别FunCat(functional catelogue)前二层的谷物蛋白质功能预测平均准确率达到85%以上,且能完成对蛋白质的第五层、第六层功能的预测; 层级结构的可回溯性使得预测结果差的功能返回至上层功能,并达到降低假阳性的概率、提高算法整体的预测准确率的效果。该研究结果可为功能类食品、药品的研发提供参考。
蛋白质;功能;预测;谷物;蛋白质语义;层级功能蛋白;蛋白质相互作用网络
0 引 言
谷物属于禾本科植物,包括小麦、大麦、燕麦、玉米、水稻、黑麦、黍稷和高粱等。谷物中含70%~72%的碳水化合物、7%~15%的蛋白质以及1%~12%的脂类[1-2]。谷物已经成为人类重要的能量来源,对健康有着举足轻重的作用。随着生活质量的提升,人们对谷物食品的功能性及保健性更加重视。食品的功能性及保健性主要依赖于食品中所含的蛋白质。因此,基于蛋白质功能的研究对功能性食品、药品的开发具有重大意义。
随着实验生物学和生物信息学的发展,大量的谷物蛋白质结构以及其功能已经被确定[3]。但仍存在着大量未经注释、功能未知的蛋白质,这些蛋白质可能蕴含可治疗或延缓人类疾病的功能。对此,谷物蛋白质的研究有利于人类对生物信息更全面地了解以及拓展生物药物研究领域,因此,目前对未知功能蛋白质的研究与预测仍是当今热点话题。
传统的预测方法通常利用蛋白质的内部结构和蛋白质序列的同源性,通过FASTA等工具在蛋白质数据库中寻找功能未知蛋白质的同源蛋白来预测蛋白质的功能[4]。但其并未考虑到蛋白质并非单一实现功能的个体,而是与其他蛋白质进行相互作用共同实现特定功能,所以使用同源性方法进行预测时会缺失部分功能。研究人员考虑到该局限性,文献[5]作者率先将蛋白质的相互作用引入到功能预测方法中,预测结果得到较好的改善。且后续的研究人员在的研究过程中对其进行了对该方法的逐渐完善[6-9]。直到蛋白质相互作用网络(protein-protein interaction networks,PPINs)的提出,使蛋白质功能预测开启了新的篇章[10]。PPINs是给定生物体的全部蛋白质所构成的网状结构,其中节点代表蛋白质、边代表两个蛋白质之间具有相互作用。根据已有的研究表明,70%~80%的蛋白质在PPINs中至少与它们直接相邻蛋白有一个共同的功能[11]。所以,从PPINs与未知蛋白质具有相互作用的蛋白功能出发,预测与已知蛋白相互作用的未知蛋白的功能成为可能[12]。
蛋白质相互作用网络逐渐发展成熟,越来越多的研究人员开始在研究中融合更多的蛋白质信息,以达到更好的预测效果。KOTLYAR[13-14]等开始使用各种蛋白质特征,将功能注释与网络拓扑结构和其他如正交学和旁系物相结合来对蛋白质的相互作用及其功能进行预测,共计得到了250 498条相互作用数据,丰富了蛋白质相互作用数据库。PAPANIKOLAOU等[15]提出了利用文本挖掘技术识别PPINs中蛋白质功能的方法。虽然这些方法都得到了大量的预测结果,但在与实际对比中发现,仍然存在大量的假阳性结果,准确率仅为60%左右。并且,这些方法都是从网络全局拓扑搜索,其时间成本过高。本文利用层级化的功能注释方案,结合蛋白质相互作用网络,使用动态贪心的策略回溯价值低的结果,使得本文提出的预测算法可以在预测大量蛋白质的同时,降低其结果的假阳性概率。
传统的方法在与相互作用网络中的蛋白质功能进行比较时,没有同时考虑蛋白质功能和功能层的语义相似度。本文利用慕尼黑蛋白质序列信息中心开发的FunCat[16]方案中不同层次的功能语义,提出一种新的蛋白质功能预测方法,该方法充分考虑了蛋白质的功能语义以及功能层次,并对具有层级特性的功能语义进行相似度的定义。基于这种新的相似度度量以及新的具有层级语义特性的蛋白质预测方法,提出一种具有更高精准度、一种更低假阳性概率等优点的算法,以期为研究人员提供便捷、准确地选择目标功能蛋白质方法,为谷物功能性食品研发与创新提供参考。
1 材料和方法
1.1 试验材料
本试验以多种谷物蛋白质作为研究对象,包括玉米、籼稻、粳稻、小麦、大豆5种常见的谷物,其蛋白质序列信息从UniProtKB/Swiss-prot数据库[17]中获取,蛋白质之间的相互作用信息从DIP(database of interacting protein)获得。目前常用的蛋白质功能注释方案为FunCat方案[16]与GO(gene ontology)术语[18]。GO术语分别从蛋白质的分子功能、细胞组分以及生物过程的角度解释蛋白质的功能及其特性[19],在探索未知的蛋白质与基因中被更多地使用;而FunCat方案从蛋白质功能语义的递进角度解释蛋白质,在探索与应用某种具体功能的情景下,其表现力优于GO注释方案。因此,本文采用FunCat功能注释方案实现谷物蛋白质的多层功能结构的预测。
FunCat是具有六层结构的功能注释策略。在表1中,展示一组FunCat术语及其所对应的功能名称实例,并描绘了FunCat的层级连接方式以及层级功能递增型结构。容易发现,在底层相同的情况下,随着层级的递增,其表达的功能更为具体。在实际的应用中,如想获取具有metabolism of the aspartate family的功能特性的蛋白质,可选择具有01.01.06及功能层级更深的01.01.06.06 等注释的蛋白质作为目标对象开展研究。由此可以快速获取目标功能所对应的蛋白质信息,减轻从海量蛋白质信息锁定目标蛋白的负担。相对于GO方案而言,FunCat方案在实际应用中更具有灵活性与快捷性,在谷物的功能性食品开发上有着重要的作用。

表1 每个级别的FunCat注释方案样本
1.2 数据转换
数据转换作为数据预处理中最重要的一个环节,是将不同的数据类型与格式进行统一。其目的是避免在试验进行中出现数据不匹配的问题,并获得试验中所需的谷物蛋白质所对应的FunCat功能表单。值得注意的一点是,FunCat最开始被设计出来的目的是为了更好描述真核单细胞生物,随着不断地完善与发展,其功能表述越来越完整,可使用范围也在不断扩大。到目前为止,尚未有谷物蛋白质FunCat注释,故无法开展FunCat对谷物蛋白质的功能预测。为了解决这一问题,本文将FunCat功能方案表单、GO术语表单以及蛋白质序列信息表单的格式进行统一(PSI-MI格式),将GO功能向FunCat功能进行转换。已知大多数的蛋白质在UniProtKB/Swiss-prot数据库中均有与之对应的GO功能表述,故可以将GO功能表单与FunCat功能表单进行功能匹配,其中约有90%的蛋白质功能可以利用描述作为关键字连接GO功能以及FunCat功能表单。剩余10%的蛋白质功能表述在GO和FunCat功能表单中并不完全一致,其原因是FunCat被定义之初用于描述和应用于真核单细胞生物,而GO被应用于全体生物,二者互不影响,以至于部分功能描述没有得到统一。为确定GO与FunCat表述不完全一致的功能描述是否为同一功能,通过遍历分别使用GO、FunCat注释的蛋白质相互作用网络、对照蛋白质相互作用信息以及缺失的FunCat功能信息,确定剩余的FunCat功能对应的GO功能。本文确定了1 360个类别的FunCat功能和7 899个谷物蛋白质具有的41 696个GO功能。部分GO功能表单与其对应的FunCat表单如表2所示,例如对于GO:0007049所对应的功能为cell cycle and dna processing,而编号为10的FunCat功能也对应于该功能,即GO功能注释方案的GO:0007049可映射为FunCat功能注释方案的10。
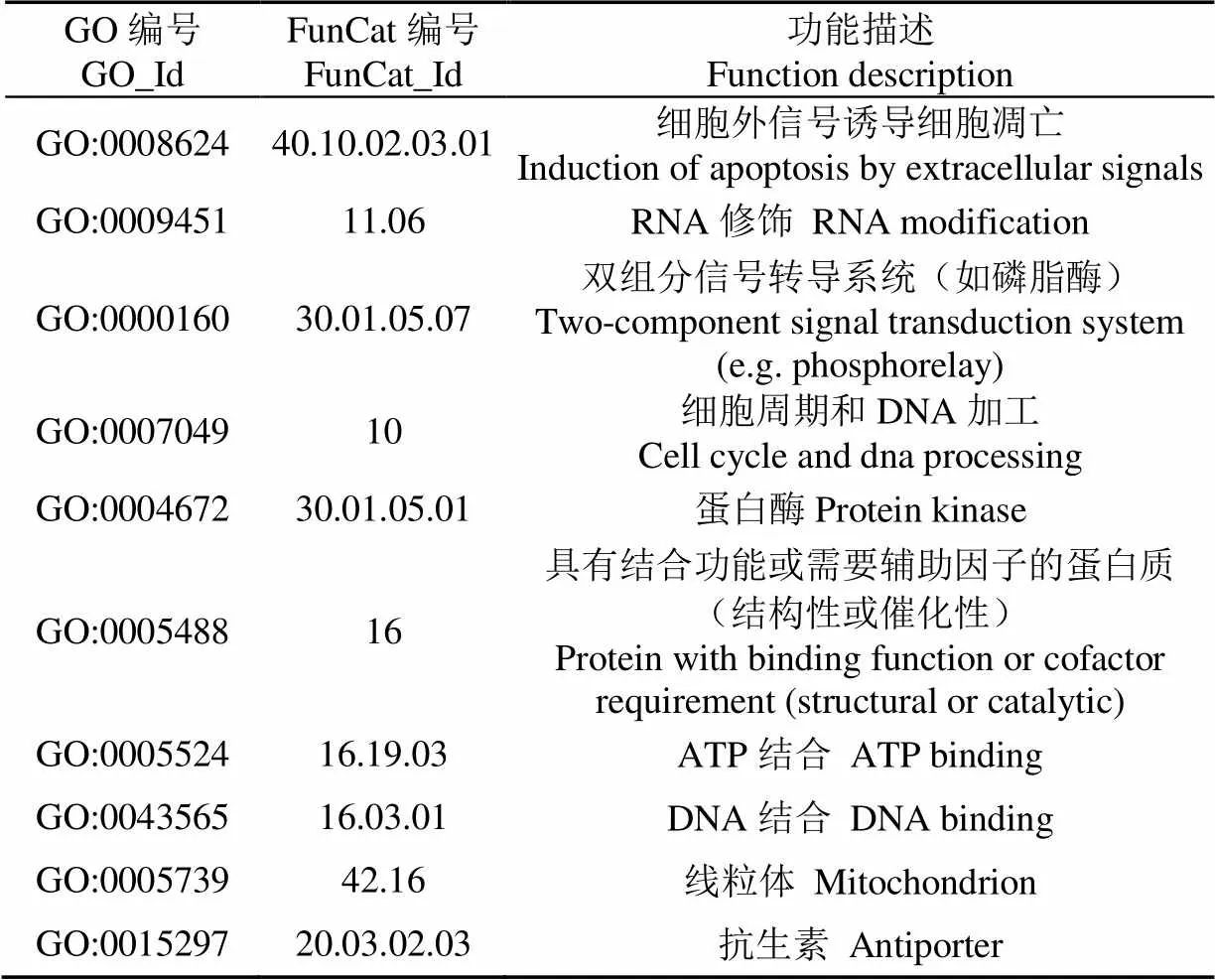
表2 FunCat与GO的数据转换表单样例
1.3 功能预测
本文首先对已有的谷物蛋白质信息构建相互作用网络,并对其网络中存在的功能信息模块进行挖掘,从而达到预测未知蛋白质功能的目的,其中最简单、有效的挖掘方法是对构建的蛋白质相互作用网络进行聚类。本文使用的多中心的非平衡-均值聚类方法[20]在实际应用中得到较好的聚类效果,试验结果表明,本文所使用的算法簇内相似度高、簇间相似度低以及其在结果的表达上优于其他聚类算法。其中,聚类过程中的聚类中心个数由轮廓系数法(silhouette coefficient)[21]确定。轮廓系数法结合了聚类的凝聚度(cohesion)和分离度(separation),其取值在[-1,1]范围内,值越大表明聚类效果越好,若某类的轮廓系数为负值则表明该类被误分。
本试验首先将所有蛋白归类到一个PPI网络;然后,使用多中心的非平衡-均值聚类方法对所构建的蛋白质相互作用网络进行聚类操作;再者,对聚类所得到的集群定义其功能特征,并记录其与未知蛋白之间的相互作用信息;最后,对未知蛋白进行功能预测。整体的功能预测流程如图1所示。
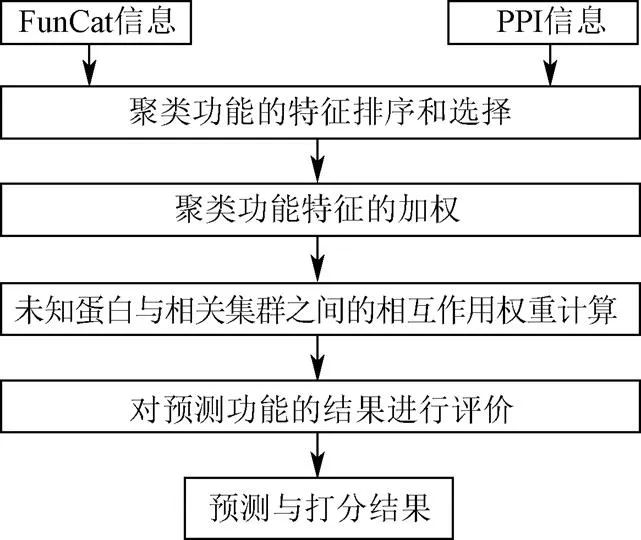
图1 谷物蛋白功能预测实现流程
1.3.1 聚类功能的特征排序和选择
网络在聚类后会形成具有模块化结构的集群。一般来说,集群的功能特征是指集群内最常见的功能可以借助FunCat的层级来计算功能出现的频率。FunCat功能方案可以将蛋白质的功能分成6个层级进行描述。利用这一特性,对一个集群中的全部蛋白质的各功能层功能进行统计计数,即可得到该集群中各层级蛋白质功能出现的频率。具体如下所示:
1)对FunCat功能中的第一层功能(以下简称一级功能)在集群中出现的频率进行排序,并对成功排序的每一个一级功能对应的第二层功能(以下简称二级功能)进行排序。以此类推,直到达到预期的水平(如第三级)。
2)选择出现频率最高的一级功能(例如选择排名前三的功能),并为每个功能对应的二级功能选出排名前三的功能,以此类推。最后,通过FunCat不同功能层级间的连接符(.)将它们连接起来形成蛋白质集群的功能特征。集群的功能特征挑选过程如图2所示:以三层功能为例(若期望更深层次的功能特征描述,可继续递进增加功能层深度)。在实例中,若01、05、02分别是第1、2、3层排名第一的功能,那么01.05.02则为该集群最显著的特征功能,其他功能特征为次显著功能特征。
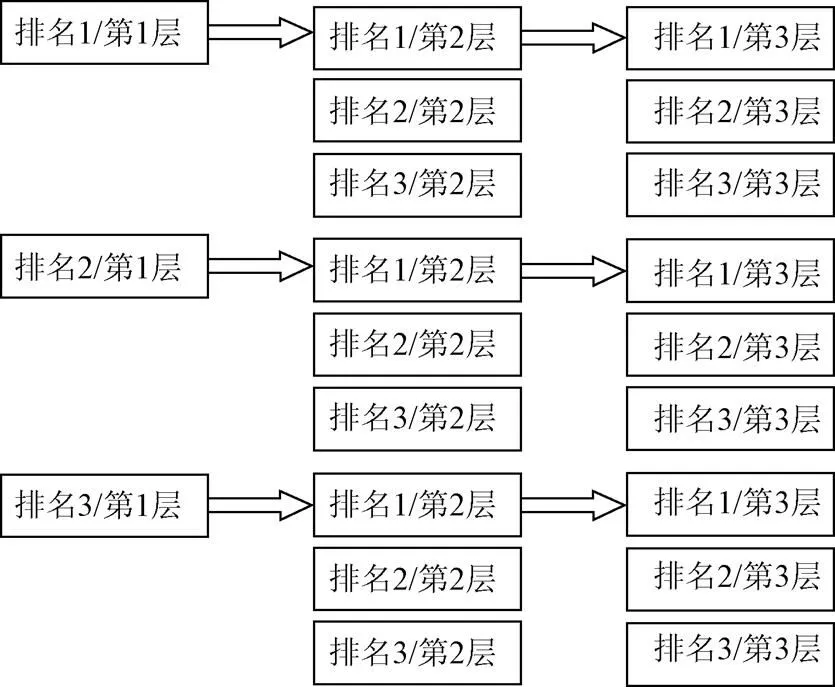
图2 集群功能特征的定义与选择流程(部分)
1.3.2 聚类功能特征加权
集群功能特征权重是用来衡量选定功能特征在集群中的重要性,是对集群内功能局部重要性的度量。假设集群的选定功能特征的数量为n,功能的层次排名为,那么功能在集群中的权重W被定义为
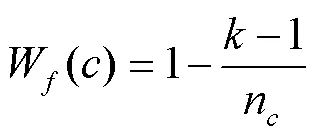
1.3.3 未知蛋白与相关集群之间的相互作用权重计算
相互作用权重是指在所有与未知蛋白质具有相互作用的集群中,各个集群对未知蛋白质功能预测的影响因子。在PPI网络中,若未知蛋白质和集群中的任何蛋白质之间存在相互作用,则认为该集群与未知蛋白质具有相互作用[22]。且后续的研究人员为衡量相互作用的权重,引入概率模型[23]。蛋白质与集群相互作用的概率u()被定义为
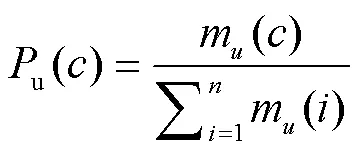
式中是未知蛋白,与未知蛋白相互作用的集群总数是,m()是未知蛋白和集群之间具有的相互作用的数量。若得到较高的概率结果,则表明集群对未知蛋白质的功能预测有较大的影响,并令未知蛋白与集群之间的相互作用概率作为相互作用权重。
1.3.4 对功能预测结果的评价与打分
未知蛋白质的功能来自与其具有较高相互作用权重的集群功能特征。因此,基于式(1)与式(2),集群投射到未知蛋白质的功能特征对功能预测的权重W被定义为
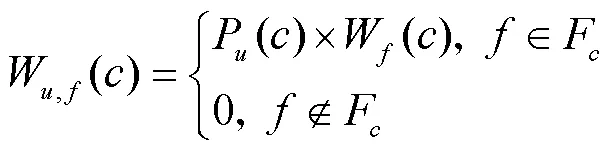
式中F为集群的功能特征集。具体运用如下:假设未知蛋白质与若干个集群具有相互作用,单个集群的3个功能特征分别为(1)10.03.01.03(2)=11.02.03.04(3)=01.03.04,根据(1) 的权重定义可得3个功能权重分别为(1)1(2)=2/3(3)=1/3;再由式(2)的相互作用概率得出(1)=3/8(2)=1/2(3)=1/8;最后通过式(3)得出该集群的3个功能与未知蛋白质之间的功能影响因子为,1(1)=3/8W,2(1)=1/4W,3(1)=1/8。
假设与未知蛋白质具有相互作用的集群数量为与具有交互作用集群的所有功能特征对未知蛋白质的功能预测得分如式(4)所示,()对应于式(3)中的集群投射到未知蛋白质的功能特征对功能预测的权重[24]。
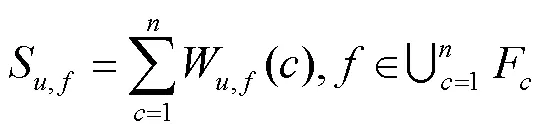
式中S是在算法模型内对预测出的蛋白质功能进行打分,其结果的大小作为确定最终蛋白质功能的一个参数,分值范围为[0,1],分值越高代表在所得到的蛋白质预测结果可能符合其真实的功能的概率越高。此过程是将模型对蛋白质功能的评判数值化,其目的是为了更好的从众多数据中挑出最有可能是“真实功能”的结果。产生的评分值不代表对模型的评估,而是算法模型对蛋白质的衡量。就该评分本身而言,需尽可能多的包含被预测蛋白质的相关权重信息[25],故将试验进程中的各步骤都进行加权包含于最终的评分公式之中,最终确定评分如式(4)。
最后,将与未知蛋白具有相互作用的集群中所有功能特征的得分进行排名,排名越靠前的功能说明越有可能是未知蛋白的真实功能。最终得到的预测功能结果可能来自多个不同的集群,这与传统的基于聚类的功能预测方法有本质区别。
1.4 评价指标的选取与性能分析
为了验证本文提出算法的有效性,在PPI数据集上基于多层功能结构的谷物蛋白质功能进行预测。本文选取精度准确率()、召回率()和值等方面进行评估。设N为所有预测的结果的的数量,N是正确预测结果的数量,N是所有已经注释的功能的数量。精度和召回率分别定义为
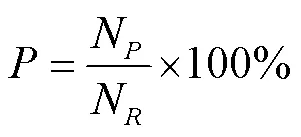
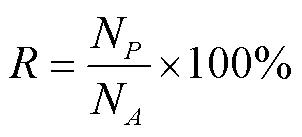
如果精度和召回率都能达到算法性能的最高值即为最佳效果。然而,通常在高精确度和高召回率之间会有一个权衡,大多情况下二者不会同时达到最好的结果。为了同时考虑这两个指标可以达到权衡最优,引入了值[26]。
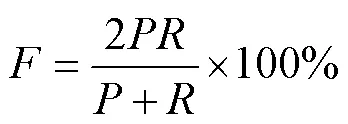
2 结果与分析
试验选取5种谷物蛋白作为研究对象,以构建大规模相互作用网络。其中籼稻、粳稻、玉米、大豆及小麦的比例分别为11.57%、53.14%、16.57%、0.42%和18.30%,其数据来源如1.1小节所述。多物种构建的蛋白质相互作用网络在进行功能性挖掘时,其结果会优于单一物种构建的蛋白质相互作用网络[27-28]。为了保证评价的客观性,从数据集中随机选择了5组数据,其中每组均由3 000个PPI数据构成,其中所包含的蛋白质作为测试集。由于PPI数据是关系型数据,故其涉及的蛋白质数目是不确定的,非定量的预测方案使得本文算法的性能更具有说服力。而对于定量测试的需求,本算法需在每组数据中随机选定若干蛋白质作为未知的蛋白质进行预测,以达到评价算法有效性的目的。
2.1 预测结果性能分析
由于FunCat是功能层级递增的结构,上一层级功能对下一层级的功能具有指导作用,所以上一级的性能评价会高于下一级的性能评价。已知现存谷物蛋白质有超过60%的功能均为三级和四级功能,五级功能和六级功能只占所有蛋白质功能的10%左右。以蛋白质实际具有的功能为标准,对本文提出的算法进行评估,对于不同层级功能的预测结果均值如表3所示,该试验为非定量的预测。可以发现,本文提出的算法在第一层级功能的精准度结果接近92%,第二层级功能的精准度达到85%左右,结果表现优于其他算法[29-31]。试验数据中约有5%的蛋白质由于没有对应的FunCat功能表达而导致预测的不成功,若将此部分功能数据剔除,所得到的结果将会得到更进一步提升。
本文提出的算法对三级功能与四级功能的准确率分别可达到78%、69%。则前四层的蛋白质功能的平均准确率高于80%。三级功能与四级功能相较于一级功能、二级功能准确率、召回率以及值均有所降低,其根本原因在于选择集群功能特征时,仅选择了排名靠前的几个功能特征,从而导致部分主要特征的丢失,并且随着预测层级的加深该特性被逐渐放大。考虑到该问题,本文将评分机制(式(4))与之结合,评分低于0.2的预测结果回溯至其上层功能记录并输出。即便没有预测至精准层级,所得的低等层级功能也对试验研究具有指导意义。该方法在一定程度上减少了预测结果的假阳性。
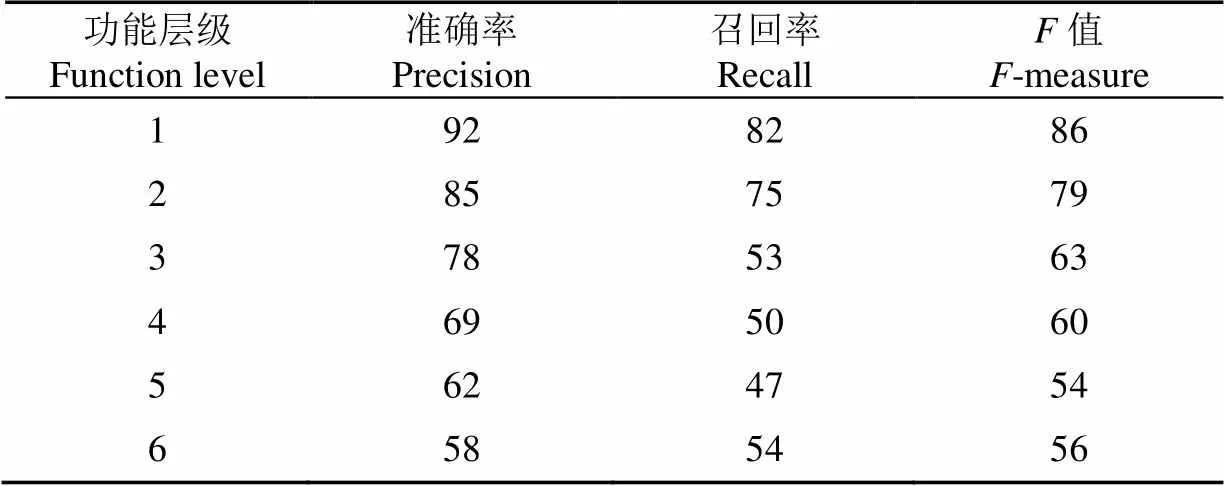
表3 谷物蛋白在FunCat不同层次的功能预测评价
为了尽可能预测出未知蛋白质的全部功能,试验过程中同时为每个集群选择多个功能特征,并在最终预测中选择多个功能得分排名较高的功能。为了验证本文所提出的方法是否可以在较大规模的未知蛋白质的情况下可以表现良好,并找到最优的规模解,采用分组的方式对其进行验证,从50个蛋白质逐步增加到400个蛋白质作为未知蛋白质样本。由于现有研究中几乎没有类似的结果可用来比较,因此,将本文提出的算法在不同大小的数据子集上对准确率、召回率和值结果进行了比较。对蛋白质功能的精准层级预测的试验结果如表4所示。特别说明,蛋白质可能拥有多个不同层级的功能,如蛋白质P86520具有的42.07、16.03.01、36.20.16分别为2级功能、3级功能、3级功能,精准层级预测需对这每个功能的最高层级功能进行预测,即2级、3级、3级功能。
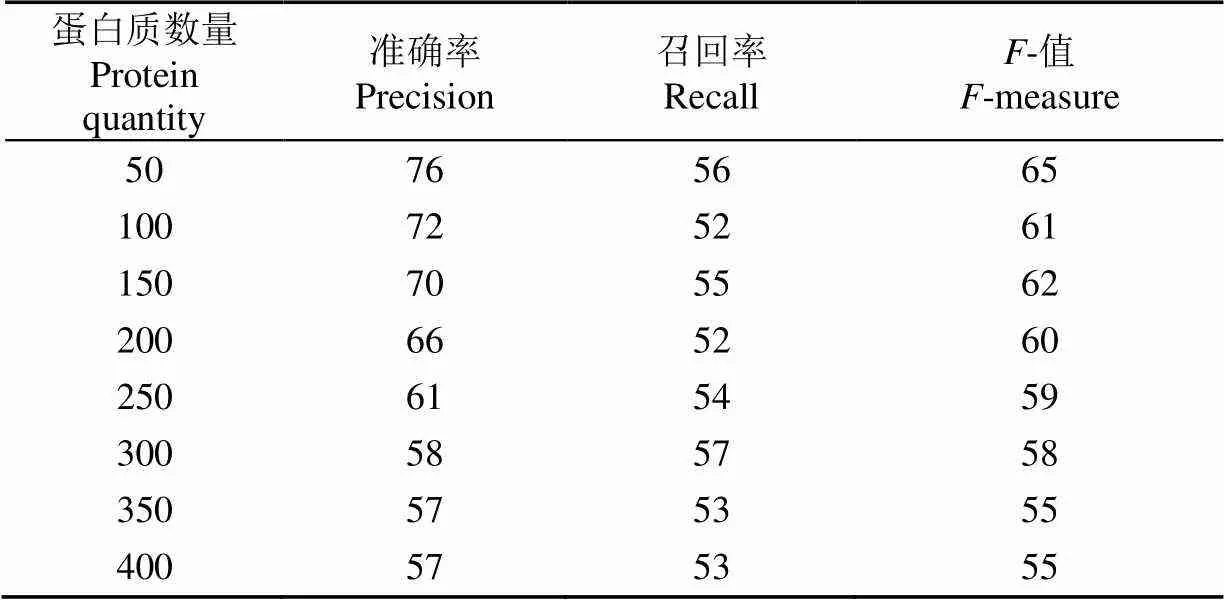
表4 对不同规模的谷物蛋白的功能预测评价
从表4可以看出,本文提出的方法对不同规模的数据集的功能预测性能相对稳定。当蛋白质数量规模取50时,取得的结果较好,其准确率可以达到76%;取100时,准确率可以达到72%;取200时,其准确率仍能达到66%,并且随着蛋白质数量规模的增大,结果趋于平缓,由此可以证明本文提出的方法具有一定的稳定性。但结果对比层级性的功能预测结果(表3),可发现指定蛋白质数目的预测性能未到达该水平,其原因在于仅在界定范围内选取排名高的功能特征,而忽视了其他功能特征,导致结果性能有所下降,但仍不可忽视其性能的优越性。
使用功能回溯后,本文提出的算法的准确率、召回率及值均有较大程度的提升,回溯前后结果对比见表5。大多高层级的功能被回溯至三级功能或四级功能。
将传统的基于FunCat功能注释方案的蛋白质功能预测方法与本文提出的方法进行对比,包括SAHA等提出的FunPred_SeqSim[29]工具、ALTUNTAS提出的DAC[30]方法、YU等[31]提出的PILL方法以及将本文提出的方法进行功能回溯后的结果进行对比。FunPred_SeqSim算法将蛋白质序列的相似性以及蛋白质相互作用信息融合预测蛋白质。DAC算法利用网络中各节点的空间信息对蛋白质的功能进行拓扑达到预测蛋白质功能的作用。PILL方法利用蛋白质功能层级的不完全层级来预测蛋白质功能的。不同算法对比结果如表5所示。
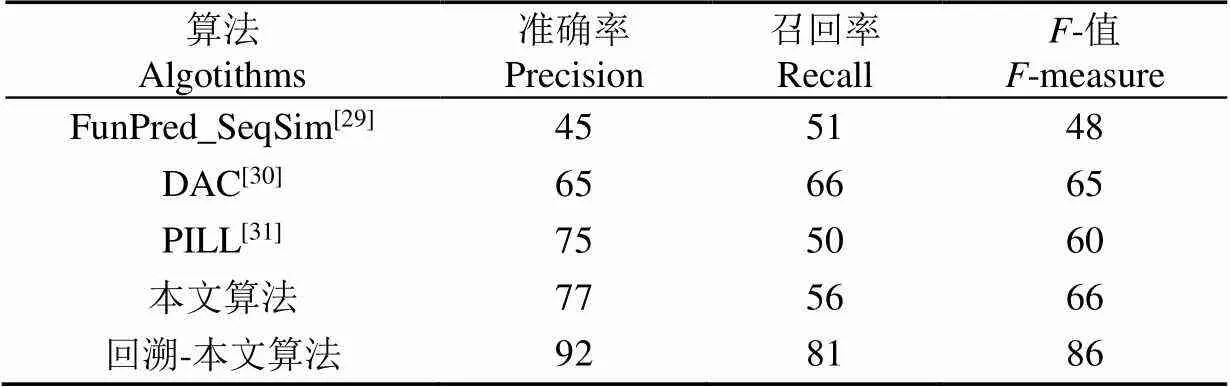
表5 本文提出的方法与其他算法的对比
从表5中可以观察到,相较于其他传统算法,本文提出的方法在准确率以及值有明显的提升且召回率也有较大改善。侧面说明本文提出的方法对于蛋白质的FunCat精准功能层级的预测有较大程度的突破,精准度可达77%;而对于模糊的蛋白质功能预测基于本文提出的方法进行功能层级的回溯还可得到更高的评价结果。模糊的蛋白质功能并不意味是错误的结果,而是锁定了蛋白质可能具有的精确功能的范围。具有功能层级越高的蛋白质越是难以准确预测,五级功能及六级功能预测的平均准确率在61%左右,召回率均值约为52%,值均值约为55%,整体水平使得预测结果不值得被信任。但将所得预测功能回溯至其上一个功能层级或两个功能层级,回溯后的蛋白质功能预测结果准确率高达92%,其结果的可信度会得到大幅提升,并且所得回溯结果可为后续研究蛋白质实际功能的生物试验提供方向,为研究人员减少一定程度的时间成本。
2.2 部分结果样本的分析与说明
在本节中,选取了几个具有代表性的结果,包含精准预测的结果以及功能回溯后的结果,如表6所示。在GO功能注释与FunCat功能注释一一对应时,可能会产生功能的多对一、一对多以及无法匹配的情况。首先,产生功能的一对多与多对一的原因是GO功能注释方案与FunCat功能注释方案均为树状注释体系,其关键节点的分枝细化程度不一,有的GO功能细化到更深的程度,而与之对应的FunCat功能没有细化到这一层次,这样就会造成GO功能的多个功能对应于FunCat功能的其中一个功能。反之,若FunCat的某一功能节点的细化程度过高,则会造成单一的GO功能对应于多个FunCat功能。对于上述的两种情况,在试验过程中选定细化程度低的一方作为试验中预测功能的标准,所得预测结果虽可能与实际功能略有偏差,但仍对试验具有指导意义。在实际的应用中,籼稻的GO:0003677与粳稻的GO:0043565对应的FunCat功能均为16.03.01,即为功能描述的多对一的情况。除此之外,还存在功能无法匹配而导致功能预测结果缺失的情况,如小麦的GO:0015066功能在FunCat功能库中没有与之对应的功能,其原因在于FunCat数据库最开始应用于细菌类以及真核单细胞生物,后续研究人员对FunCat数据库进行扩充时还没有完全丰富该库,但预留的第99类可作为扩充的信息源。
在试验过程中,层次递增型的预测模型可能会导致获取过度预测结果或降维预测结果(过度预测为超越蛋白质实际功能所具有的层级;降维预测为未达到蛋白质实际功能所具有的层级)。例如,籼稻的GO:0004674对应的功能为30.01.05.01.06,但在实际回溯后预测的结果中得到的是30.01.05.01,这是由于30.01.05.01.06是30.01.05.01的分枝功能,后者功能包含前者,故认为所得预测结果正确。而大豆的GO:0005783功能被预测成42.07.01,但其实际功能为42.07,是过度预测结果,本文认为过度预测的结果是不正确的,即使其结果打分为0.96,但仍应从结果中删除。考虑到过度预测结果过多会影响研究人员在实际应用中对蛋白质功能的判断,因此要减少此类情况的出现,所以在进行集群特征选择时将权重过低的功能特征从特征集中删除。
由试验结果可知,虽试验结果有较小的偏差,但仍有95%以上蛋白质被注释,并在注释的蛋白质中被正确注释的达到90%以上,整体准确率可达80%以上。未被注释的约5%的蛋白质是由于FunCat功能库的缺失,无法匹配功能,导致在预测时无法被成功注释。
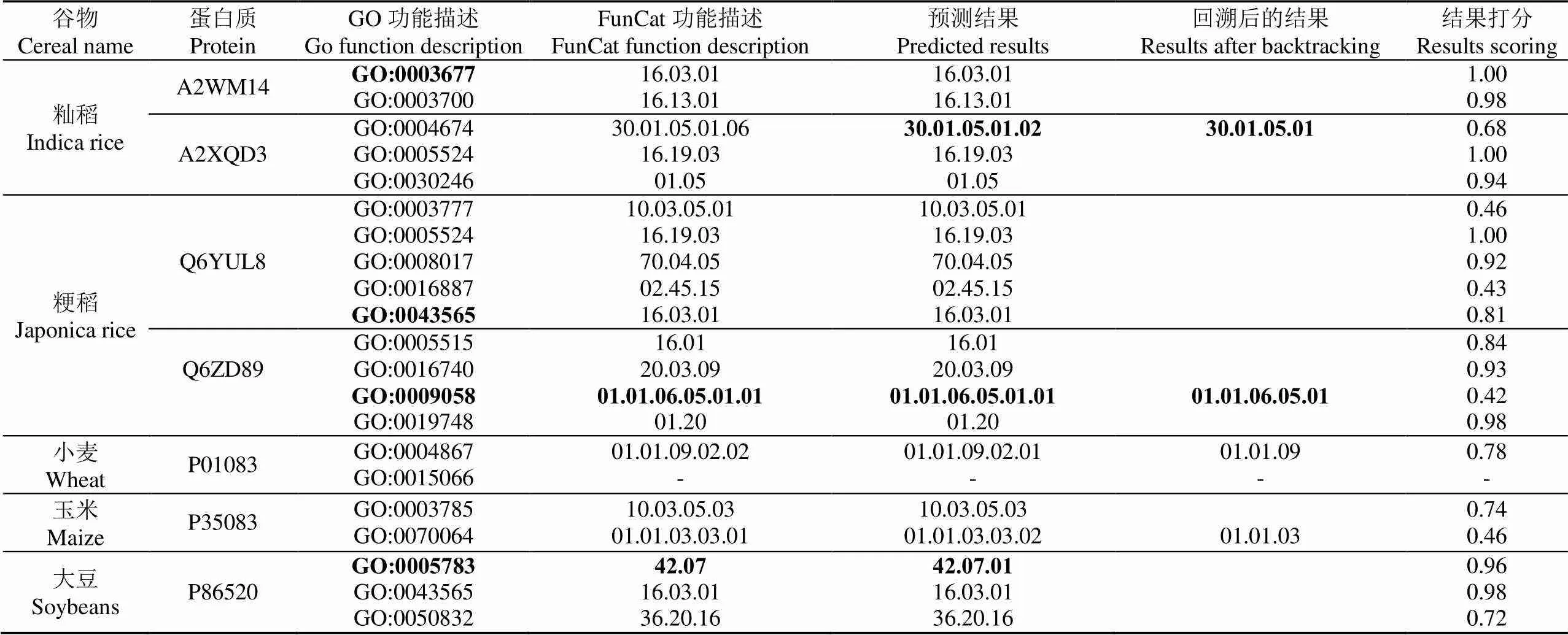
表6 谷物蛋白质功能预测结果部分样例
3 结论与讨论
本文将具有FunCat层级结构的蛋白质功能预测应用于谷物蛋白中,并提出了利用蛋白质语义及分层蛋白质功能结构的功能预测框架。该方法可以在预测谷物蛋白质功能时指定预测蛋白质的功能层级,并且可回溯功能的预测方法使得假阳性的结果大幅度降低。
通过主流的评价方法对本文所提出的方法进行评价,证明该方法对谷物蛋白质的功能预测有良好的表现,对于蛋白质的功能预测准确率约为77%,而对于回溯后的模糊的蛋白质功能预测准确率可以达到92%。因此,该方法不仅可以预测蛋白质的精准功能,还可以预测蛋白质功能范围,为研究人员在功能性蛋白质选择时提供便利。
试验结果表明,本文提出的方法在预测大量未知蛋白质功能方面是有效并且可行的。但对于界定了具体范围的蛋白质数据集中进行预测,其准确率和召回率结果不佳。其主要原因是,在试验集群的特征选择时仅选取了权重及评分靠前的几个特征功能,但实际应用中其他影响权重相对低的功能特征仍有可能成为未知蛋白的功能组成。
尽管本文提出的方法在谷物数据集上表现良好,但仍有一些问题可以在未来进行改进。目前,试验中使用的相似性度量是基于直接与未知蛋白质具有相互作用的蛋白质的功能。然而,仅通过直接相互作用的蛋白质功能很难获取一个未知蛋白质的全部功能,未知蛋白质也可能与其他没有直接相互作用的蛋白质共享功能。那么如何将PPI(protein-protein interaction)中的这种非直接相互作用的功能成功预测可作为下一步研究的内容。
[1] XU Y, YANG J, DU L, et al. Association of whole grain, refined grain, and cereal consumption with gastric cancer risk: A meta‐analysis of observational studies[J]. Food Science & Nutrition, 2019, 7(1): 256-265.
[2] 张敏,吴崇友,陈旭,等.近红外光谱式联合收割机谷物蛋白质含量检测系统设计[J].农业工程学报,2021,37(1):36-43.
ZHANG Min, WU Chongyou, CHEN Xu, et al, Design of near-infrared spectral grain protein detection system for combine-harvesters[J]. Transactions of the Chinese Society of Agricultural Engineering (Transactions of the CSAE),2021,37(1): 36-43.(in Chinese with English abstract)
[3] GONG X, AN Q, LE L, et al. Prospects of cereal protein-derived bioactive peptides: Sources, bioactivities diversity, and production[J]. Critical Reviews in Food Science and Nutrition, 2022, 62(11): 2855-2871.
[4] ISLAM S I, JAHAN M M. Functional annotation of uncharacterized protein from photobacterium damselae subsp. piscicida () and comparison of drug target between conventional medicine and phytochemical compound against disease treatment in fish: An in-silico approach[J]. Genetics of Aquatic Organisms, 2022, 6(3): 1-14.
[5] WALHOUT A J M, SORDELLA R, LU X, et al. Protein interaction mapping in C. elegans using proteins involved in vulval development[J]. Science, 2000, 287(5450): 116-122.
[6] VELLA D, MARINI S, VITALI F, et al. MTGO: PPI network analysis via topological and functional module identification[J]. Scientific Reports, 2018, 8(1): 5499.
[7] ZHOU X, ZHENG W, LI Y, et al. I-TASSER-MTD: A deep-learning-based platform for multi-domain protein structure and function prediction[J]. Nature Protocols, 2022, 17(10): 2326-2353.
[8] 雷秀娟,高银,郭玲. 基于拓扑势加权的动态PPI网络复合物挖掘方法[J]. 电子学报,2018,46(1):145-151.
LEI Xiujuan, GAO Yin, GUO Ling. Mining protein complexes based on topology potential weight in dynamic protein-protein interaction networks[J]. Chinese journal of electtonics, 2018, 46(1): 145-151. (in Chinese with English abstract)
[9] PENG W, WANG J, CAI J, et al. Improving protein function prediction using domain and protein complexes in PPI networks[J]. BMC Systems Biology, 2014, 8(1): 1-13.
[10] ROSA S, BERTASO C, PESARESI P, et al. Synthetic protein circuits and devices based on reversible protein-protein interactions: An overview[J]. Life, 2021, 11(11): 1-10.
[11] STATELLO L, GUO C J, CHEN L L, et al. Gene regulation by long non-coding RNAs and its biological functions[J]. Nature Reviews Molecular Cell Biology, 2021, 22(2): 96-118.
[12] BAN Z, YUAN P, YU F, et al. Machine learning predicts the functional composition of the protein corona and the cellular recognition of nanoparticles[J]. Proceedings of the National Academy of Sciences, 2020, 117(19): 10492-10499.
[13] KOTLYAR M, PASTRELLO C, PIVETTA F, et al. In silico prediction of physical protein interactions and characterization of interactome orphans[J]. Nature Methods, 2015, 12(1): 79-84.
[14] CHEN K H, WANG T F, HU Y J. Protein-protein interaction prediction using a hybrid feature representation and a stacked generalization scheme[J]. BMC Bioinformatics, 2019, 20(1): 1-17.
[15] PAPANIKOLAOU N, PAVLOPOULOS G A, THEODOSIOU T, et al. Protein–protein interaction predictions using text mining methods[J]. Methods, 2015, 74: 47-53.
[16] MEWES H W, DIETMANN S, FRISHMAN D, et al. MIPS: analysis and annotation of genome information in 2007[J]. Nucleic Acids Research, 2008, 36(suppl_1): 196-201.
[17] BOUTET E, LIEBERHERR D, TOGNNOLLI M, et al. UniProtKB/Swiss-Prot, the manually annotated section of the UniProt KnowledgeBase: How to use the entry view[J]. Plant Bioinformatics: Methods and Protocols, 2016, 1374(1): 23-54.
[18] Gene Ontology Consortium. The gene ontology resource: 20 years and still GOing strong[J]. Nucleic Acids Research, 2019,47(D1): D330-D338.
[19] KANEHISA M, SATO Y. KEGG Mapper for inferring cellular functions from protein sequences[J]. Protein Science, 2020, 29(1): 28-35.
[20] 亓慧. 多中心的非平衡K-均值聚类方法[J]. 中北大学学报(自然科学版),2015,36(4):453-457.
QI Hui. Imbalanced K-means clustering method with multiple centers[J]. Journal of North Central University (Natural Sciences Edition), 2015, 36(4): 453-457. (in Chinese with English abstract)
[21] DINH D T, FUJINAMI T, HUYNH V N. Estimating the optimal number of clusters in categorical data clustering by silhouette coefficient[C]//International Symposium on Knowledge and Systems Sciences. Springer, Singapore, 2019: 1-17.
[22] HUTTLIN E L, BRUCKNER R J, NAVARRETE-PEREA J, et al. Dual proteome-scale networks reveal cell-specific remodeling of the human interactome[J]. Cell, 2021, 184(11): 3022-3040.
[23] CHATTLA S, SHMUELI G. Linear probability models (LPM) and big data: The good, the bad, and the ugly[J]. Indian School of Business Research Paper Series, 2016(11): 1-45.
[24] 李峰,孙波,王轩,等. 层次分析法结合熵权法评估农村屋顶光伏系统电能质量[J]. 农业工程学报,2019,35(11):159-166.
LI Feng, SUN Bo, WANG Xuan, et al. Power quality assessment for rural rooftop photovoltaic access system based on analytic hierarchy process and entropy weight method[J]. Transactions of the Chinese Society of Agricultural Engineering (Transactions of the CSAE), 2019, 35(11): 159-166. (Transactions of the CSAE)
[25] MATEO J R S C. Weighted Sum Method and Weighted Product Method[M]//Multi criteria analysis in the renewable energy industry. Springer, London, 2012: 19-22.
[26] HAND D,CHRISTEN P. A note on using the F-measure for evaluating record linkage algorithms[J]. Statistics and Computing, 2018, 28(3): 539-547.
[27] STRYDOM T, CATCHEN M D, BANVILE F, et al. A roadmap towards predicting species interaction networks (across space and time)[J]. Philosophical Transactions of the Royal Society B, 2021, 376(1837): 1-17.
[28] VAN Leene J, HAN C, GADEYNE A, et al. Capturing the phosphorylation and protein interaction landscape of the plant TOR kinase[J]. Nature Plants, 2019, 5(3): 316-327.
[29] SAHA S, CHATTERJEE P, BASU S, et al. Multiple functions prediction of yeast saccharomyces cerevisiae proteins using protein interaction information, sequence similarity and FunCat taxonomy[C]//2020 IEEE 1st International Conference for Convergence in Engineering (ICCE). IEEE, India, Kolkata, 2020: 170-174.
[30] ALTUNTAS V. Diffusion alignment coefficient (DAC): A novel similarity metric for protein-protein interaction network[J]. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 2022(6): 1-11.
[31] YU G, ZHU H, DOMENICONI C. Predicting protein functions using incomplete hierarchical labels[J]. BMC Bioinformatics, 2015, 16(1): 1-12.
Prediction of cereal protein function based on multilayer functional structures
SHEN Tingting1, LIU Jing1※, GUAN Xiao2,3
(1.,,201306,; 2.,,200093,;3.(),200093,)
Cereals are very valuable food sources of healthy and sustainable protein. Food innovations in cereal protein are ever transitioning to more sustainable food systems for healthy diets. A more precise understanding is required by the functions that cereal proteins have. The application of cereal proteins has greatly contributed to genomics and food science today. In this study, a functional prediction was proposed for the cereal proteins using a multilayer functional structure, in order to select the functional proteins more conveniently and accurately. A large-scale interaction network was also constructed with the indica, japonica, wheat, maize, and soybean data. Firstly, the relevant functions of unknown proteins were explored via the interaction of functional features of clusters with the unknown proteins. Secondly, new protein weights, semantic similarity, and functional hierarchy weights were defined to determine the possible functions of proteins. Finally, the grain protein function was further determined using a scoring mechanism in the prediction of the function. The results show that better performance was achieved to predict the function of cereal proteins, particularly with a precision of about 77% for the accurate protein function prediction and up to 92% for the fuzzy protein function prediction using retraceability. A great contribution was made to determine the functional range of unknown proteins, especially with the high efficiency of prediction. The precision of protein function prediction varied significantly at different levels, with an average precision of 92% at level-1, 85% at level-2, and 69% at the level-4. More importantly, the average precision was close to 80% in all six levels of FunCat. As such, the multi-layer functional structure of proteins was predicted to calculate the number of unknown proteins with different sizes. The precision of the prediction was 76% at an unknown protein size of 50, 72% at an unknown protein number of 100, and 66% at an unknown protein number of 200. There was no sharp decrease with the significant increase in the prediction size. It infers that the prediction still performed the best in the case of large-scale unknown proteins. A comparison was made with the latest algorithms, such as FUNPRED_SEQSIN, DAC (Diffusion Alignment Coefficient), and PILL (Predict protein function using Incomplete hierarchical LabeLs). In terms of precision, recall, and F-measured, the performance of the improved prediction was significantly better than the others. The experimental results show that 1) the prediction can be expected to serve as the predicted function hierarchical, particularly for the protein with the specified function, or the available protein functions of specified functional levels; 2) The average precision of the cereal protein function in the first four layers of FunCat (Functional Catelogue) can reach more than 80%, even to realize the prediction of the fifth and sixth layers of the protein; 3) The retrospective nature of the hierarchy can allow the functions with the low predictions to be returned to the higher level functions. As such, the probability of false positives was reduced to improve the overall prediction accuracy. The finding can also provide a strong reference to the protein function prediction in the food industry.
protein; function; prediction; cereals; protein semantics; hierarchical functional proteins; protein-protein interaction network
10.11975/j.issn.1002-6819.202210046
TP391.4
A
1002-6819(2023)-01-0261-08
沈婷婷,刘静,管骁. 基于多层功能结构的谷物蛋白质功能预测[J]. 农业工程学报,2023,39(1):261-268.doi:10.11975/j.issn.1002-6819.202210046 http://www.tcsae.org
SHEN Tingting, LIU Jing, GUAN Xiao. Prediction of cereal protein function based on multilayer functional structures[J]. Transactions of the Chinese Society of Agricultural Engineering (Transactions of the CSAE), 2023, 39(1): 261-268. (in Chinese with English abstract) doi:10.11975/j.issn.1002-6819.202210046 http://www.tcsae.org
2022-10-08
2022-11-21
国家自然科学基金项目(32172247);内蒙古自治区科技重大专项“燕麦新品种选育、绿色栽培技术与营养功能产品研究与示范”(2021ZD0002)
沈婷婷,研究方向为生物信息、机器学习。Email:shentt_2021@qq.com
刘静,博士,副教授,研究方向为生物信息、信息技术与食品功能交叉领域的研究。Email:jingliu@shmtu.edu.cn
