基于序列的蛋白质二面角预测研究综述
2023-03-27郑美丽张步忠
郑美丽,朱 琪,张步忠,2
(1.安庆师范大学 计算机与信息学院,安徽 安庆 246013;2.苏州大学 江苏省计算机信息处理技术重点实验室,江苏 苏州 215006)
0 引 言
蛋白质是由氨基酸缩水链接成的一种有机复合物,一个氨基酸残基的基本构成有中心Cα原子、氨基(-NH2)、羧基(-COOH)、氢键(-H)和侧链R基团。蛋白质三维结构中,骨架主链上的二面角很大程度上反映了三维构象。一个氨基酸残基通常对应两个二面角(首尾残基除外)[1],φ(phi)和ψ(psi),范围在-180°至180°之间。围绕N-Ca的C-N-Ca-C原子构成φ二面角,围绕Ca-C键的N-Ca-C-N构成ψ二面角,实例如图1所示。蛋白质主链二面角(φ,ψ)是蛋白质结构的一部分,研究蛋白质二面角对于蛋白质功能的研究具有重要的意义。诺贝尔奖获得者Anfinsen[2]的实验表明,蛋白质的结构信息包括二面角蕴含于其序列之中,从而表明从序列出发进行蛋白质二面角预测是可行的。
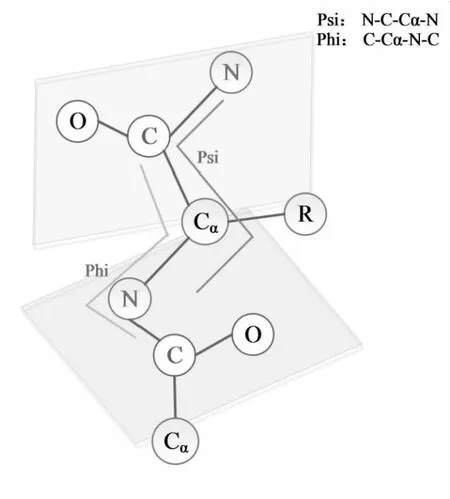
图1 蛋白质二面角
骨架二面角作为一种重要的结构约束,它对主链构象变化的影响远大于键长和键角,在蛋白质结构预测的空间进行采样以研究蛋白质折叠和细化中起着关键作用,准确预测骨架二面角可以加速对低能结构构象空间的有效采样,大大推进三级结构预测。
1 特征表示和数据集
1.1 序列表示
蛋白质序列残基的表示,主要有位置特异性评分矩阵[3](PSSM)、隐马尔可夫模型打分矩阵[4](HMM)、物理化学性质[5](PP)、蛋白质二级结构[6](SS)、溶剂可及性[7](SA)、序列编码[8](SC)等。
1.1.1 位置特异性评分矩阵
序列进化信息对揭示蛋白质结构和功能非常重要。多序列比对方法PSI-BLAST[9]产生的位置特异性评分矩阵能揭示序列进化信息,被广泛应用在蛋白质相关的生物信息学中。如式(1)所示,PSSM是形如L×20的矩阵,其中L是蛋白质序列长度,行是序列中残基,列是现有20种氨基酸。P(i→j)表示序列中第i个残基突变为第j种氨基酸残基的概率。对于将PSSM数据作为序列表示,还需进行归一化处理。
(1)
1.1.2 隐马尔可夫模型评分矩阵
在多序列比对中,HH-suite[10]套件中的HHblits基于其专用格式的多序列数据库,通过聚类UniProt或者NR库,将序列长度对齐性80%以上、相似度20%以上的序列聚集,并生成对应的HMM特征文件。通过HHsearch/HHblits生成的HMM格式中,HMM数据部分表示的是该位置残基向对应残基变异的发生概率,用-1 000*lb(frequency)表示成正整数,“*”表示零。随后的一行是10种转移概率。在作为数据特征表示时,可取前30列。
1.1.3 氨基酸的物理化学特性
蛋白质其物理化学性质一部分与氨基酸相似,一部分在特定环境下具有特定的性质。在蛋白质结构特性预测中,常用的氨基酸理化性质有:空间参数、极化率、体积、疏水性、等电点、螺旋概率和片概率,具体取值参见文献[5]。
1.1.4 序列编码
蛋白质一级序列是字母编码,对一级序列常用0-1编码表示,多数用21或22维向量的正交编码。由于该编码形式只有一个非零向量,不利于梯度优化类算法值更新,Zhang[11]采用自编码器方式将0-1稀疏向量映射到稠密向量,计算方法如式(2),用h表示新的编码。
(2)
1.1.5 其 它
蛋白质结构决定功能,描述其空间特性的二级结构、溶剂可及表面积、残基接触图[12](Contact Map,CM)、多序列比对信息(MMseqs2)等也应用到了二面角预测中。OPUS-TASS和OPUS-TASS2方法还使用独特的PSP[13](Potential Based On Side Chain Packing)特征。为方便对比,表1列出了近年来的典型预测算法的特征表示。

表1 预测方法的输入特征
1.2 输 出
蛋白质二面角预测值表示主要有:数值(已归一化),二面角的正弦和余弦函数值。SPINE X、Real-SPINE、Real-SPINE 2.0、Real-SPINE 3.0、DANGLE等方法输出都是将二面角进行归一化。SPIDER 2、SPIDER 3、SPIDER3-Single、DeepRIN、RaptorX-Angle、SPOT-1D、ProteinUnet、CRRNN2、SPOT-1D-single等通过输出二面角的正弦、余弦函数来消除角度周期性,再通过公式α=tan-1[sinα/cosα]还原二面角。DESTRUCT、ANGLOR则是直接输出二面角的角度。

1.3 评价指标
二面角预测评价标准有皮尔逊相关系数(PCC)、平均绝对误差(MAE)、均方根误差(RMSE)等。在进行蛋白质二面角预测评价时,对预测值P'和真实值E之间的差值通常先按式(3)将二面角进行角度变换,其中P'是预测二面角的原始值。PCC、MAE、RMSE分别通过式(4)~(6)计算。
(3)
(4)

(5)
(6)
1.4 数据集
1.4.1 训练集
训练集多数来自PISCES CullPDB[33]挑选的数据集,序列之间的相似度一般低于30%。也常从PDB蛋白质数据库中直接抽取序列并筛选用作训练集。
1.4.2 测试集
对模型泛化性能进行测试用的数据集,多数来自一些公开数据集,如CB513、CASP(10-14)数据集等。另一种是算法提出者给出的测试集合,如TEST2016、TEST2018[27]、CASP-FM56[34]等。表2和表3分别给出了近年来的预测方法对(φ,ψ)二面角的预测性能,测试数据用CASP公共数据集。“FM”表示数据集中是无模板序列,预测难度更大。对比指标用PCC和MAE,“-”表示指标缺失。

表2 φ角在各测试集的评价指标
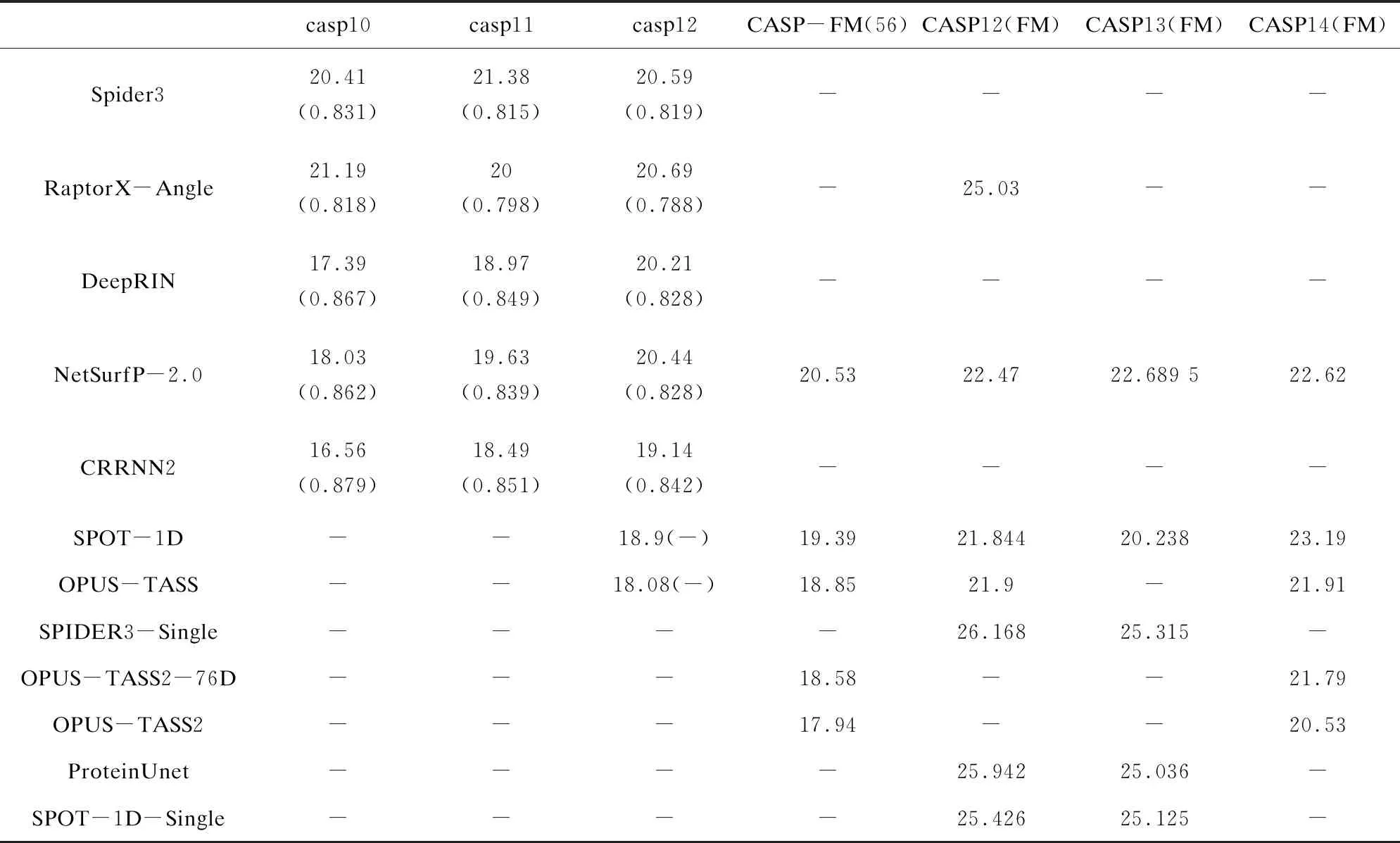
续表2

表3 ψ角在各测试集的评价指标
ψ角的预测难度高于φ角。从预测结果看,自Spider 3起的深度学习方法均取得了更优性能,一方面是训练数据更多,另外一方面参数更多的深度学习方法得到充分训练后,泛化性能更好。特别地,OPUS-TASS2性能最好,其次分别是OPUS-TASS和SPOT-1D。这三个方法都是参数规模大的集成模型,SPOT-1D和OPUS-TASS混合使用长短期记忆网络[35](Long-Short Term Memory,LSTM)、卷积神经网络[36](Convolutional Neural Networks,CNN)和残差网络[37](Residual Neural Network,ResNet);OPUS-TASS2进一步融合Transformer[38]。SPOT-1D和OPUS-TASS2都将Contact map作为输入。
2 二面角预测
二面角预测可归类为回归问题,随着蛋白质已知结构的数据集增多,机器学习方法已应用到该问题中。
2.1 传统机器学习方法
二面角预测引入到计算领域最初是作为辅助手段提升二级结构预测性能。2000年Bystroff[39]提出用隐马尔可夫模型预测二面角构象,将(φ,ψ)映射到10个区域和1个顺式肽结构。2004年,Kuang等[40]用支持向量机和神经网络预测二面角构象,将(φ,ψ)映射到(A,B,G,E)四个区域。
2005年Wood等[14]提出DESTRUCT方法,用级联反馈输入策略构建三个神经网络,预测二级结构和ψ二面角,但其关注点依然在二级结构。2008年,Wu和Zhang[15]提出ANGLOR方法,用PSSM和计算软件预测的二级结构、溶剂可及性作为输入,用神经网络预测φ角、支持向量机预测ψ角。2009年Shen等[41]提出TALOS+模型,利用两级前馈神经网络预测(φ,ψ)二面角。
2012年Song等[17]提出TANGLE,用两阶段的支持向量回归(SVR)策略预测骨架二面角。TANGLE不依赖已知结构信息,输入是PSSM和PSIPred软件预测的二级结构、Scratch预测的溶剂可及性、Disopred2预测的固有无序信息、序列长度以及序列权重,输入到第一阶段SVR后,预测结果再输入第二阶段的SVR。Song还综合验证了输入窗口尺寸和输入特征组合对性能的影响。TANGLE完全从一级序列出发以及两阶段训练、逐步求精的策略,对后续研究工作有深刻的影响。
2010年,Cheung等[42]提出DANGLE模型,用贝叶斯生成模型预测二面角。利用残基键化学位移特性,将(φ,ψ)二面角映射到36×36的10°方格拉氏空间(Ramachandran Space)图中。利用贝叶斯公式计算查询散布模式(Query Scatter Pattern,QSP)下的(φ,ψ)概率。
限于计算资源和已知蛋白质三维结构不多等因素,传统计算方法性能有待改进。但给本问题研究提供了参考意义。
2.2 深度学习方法
近年来,深度学习方法由于其良好的泛化性能,广泛应用在多个领域。2014年,Lyons等[43]用深度稀疏自编码器堆叠的模型SPIDER预测骨架Cα上的θ和τ二面角。2015年Heffernan等[21]提出的SPIDER2可以预测蛋白质二级结构、二面角和溶剂可及表面积。SPIDER2模型结构和SPIDER一样,在训练时进行三次迭代,上一次训练结果作为下一次迭代训练的输入。实验表明迭代式训练能有效提升模型泛化性能。
2017年,Li等[44]将受限的玻尔兹曼机(RBM)与深度神经网络结合,设计出了深度递归RBM(DReRBM)模型。在受限的玻尔兹曼机的基础上,将上一次的输出ht-1作为本次输入,充分拟合了蛋白质序列上下游环境。DReRBM模型由输入层、隐藏层和输出层组成,多个RBM堆栈在其中,以一种逐步的方式训练,一个训练过的RBM的隐藏数据作为可见的输入数据馈送给下一个RBM,模型梯度计算通过吉布斯采样完成。
2017年,Heffernan等[22]提出SPIDER3模型预测蛋白质二级结构、溶剂可及性、接触图和二面角。SPIDER3由两层双向长短期记忆网络[45](BLSTM)构建。SPIDER3和SPIDER2一样,采用迭代训练的策略,上一轮输出作为下一轮输入,共迭代了4次。SPIDER3分别训练了回归和分类模型。BLSTM能记忆前向和后向两个方向的时序输入信息,较好地拟合了蛋白质序列残基和左右上下文相关的特性,能够学习距离较远和距离较近的序列内的依赖关系。
卷积神经网络[36]结合残差网络[37],通过网络层加深提升长范围特征感知。2018年,Gao等[24]提出了RaptorX-Angle模型。RaptorX-Angle中堆叠了多个残差块。结合K-means算法,首先,从训练数据中生成一组(φ,ψ)的聚类,从中可以得到每个聚类的分布;然后,利用深度学习方法对离散标签进行预测;最后,通过混合经验聚类及其预测概率来预测实际二面角值。
另一典型CNN结构模型是Fang等[25]在2018年提出的DeepRIN。DeepRIN结合Inception ResNet,构建残差Inception块,并堆叠两层残差Inception块。DeepRIN用小窗口卷积来提高网络的计算效率。DeepRIN使用9 000条训练数据,并将每条序列长度对齐到700,输入不再使用窗口形式。
鉴于RNN在长范围特征获取的优势、CNN局部特性获取和ResNet便捷残差传递的特点,多数模型结合三者用于预测二面角。2019年Klausen等[28]提出了NetSurfP-2.0模型,预测残基的溶剂可及性、二级结构、无序蛋白和骨架二面角。NetSurfP-2.0输入是HMM特征和序列编码,分别经过32个卷积核129、257的CNN后,输入两层BLSTM网络,BLSTM单向1 024个单元。NetSurfP-2.0使用了10 337条训练序列,其参数规模也达到了3 400万,过多网络参数给训练和预测带来了不便。
2019年,Kim等[46]提出使用生成对抗网络(GAN)进行二面角预测,训练了GAN的鉴别器来估计密度,但模型的显式密度不易处理。因此,引入噪声对比估计(Noise-Contrastive Estimation,NCE)来估计非归一化统计模型的归一化常数,即引入了噪声对比估计生成对抗网络(NCE-GAN),通过从已知分布(如噪声对比估计)中输入噪声样本,并为鉴别器添加相应的类,从而实现生成对抗网络的显式密度估计。
2019年,Hanson等[27]提出SPOT-1D模型,预测蛋白质二级结构、二面角、溶剂可及性和残基接触数(Contact Number)。SPOT-1D的输入在PSSM、HMM特征和理化性质的基础上,将预测的接触图作为输入改进模型泛化性能。SPOT-1D利用BLSTM和ResNet混合模型的集成来识别和传播整个序列的短期和长期依赖,SPOT-1D由9个网络结构的模型集成。SPOT-1D训练集包含10 029条序列,分别训练了分类和回归两类模型,总模型文件大小10 GB左右。SPOT-1D在多个任务上均取得较好性能,但其庞大的模型不利于在生物领域应用开展。
2020年,Xu等[31]提出的OPUS-TASS性能比SPOT-1D更好。OPUS-TASS输入为PSSM、HMM、理化性质和19位PSP,分别送到5层CNN网络、2层Transformer网络(编码部分),两部分合并得到228维的数据再送给4层BLSTM网络。OPUS-TASS分别集成7个模型用于分类和回归预测。OPUS-TASS模型文件3.7 GB,比SPOT-1D要小,但依然对实际应用的资源要求较高。
2018年,Heffernan等提出了仅使用序列信息的SPIDER3-Single[23]模型,SPIDER3-Single的网络结构和训练方法与SPIDER3类似,不同的是仅用了20维序列编码作为输入。但SPIDER3-Single模型泛化性能和SPIDER3相比还是有较大差距。2021年Kotowski等[29]提出ProteinUnet模型,ProteinUnet输入和SPIDER3-Single一样,但大幅度提升了预测性能。
2021年,Singh等[30]同样提出了面向单序列输入的SPOT-1D-Single模型。SPOT-1D-Single集成了三个不同的结构模型,也分别面向分类和回归训练。SPOT-1D-Single使用39 120条训练序列。SPOT-1D-Single泛化性能不如SPOT-1D,但已超越ProteinUnet,并已有了一定的实用价值。
3 展 望
通过上述分析,计算方法特别是深度学习预测二面角,取得了较好进展,但预测性能依然有提升空间,针对相关研究,可以从以下几个方面进行思考:
(1)单序列输入更方便生物学人员使用。现有模型多依赖PSSM、HMM等多序列比对信息,对非专业人员要求高。仅有序列编码信息的单序列模型,对生物学人员更友好,而单序列输入模型性能还有待提高。
(2)需要设计对计算资源依赖更少的模型。在泛化性能一致时,轻量级模型更方便用户使用,推理时对计算资源依赖更少。
(3)可将多个问题联合解决。二面角、二级结构、接触图等蛋白质结构层面问题,相互依赖。如二级结构预测性能提升,同样能推动二面角预测性能。将预测得到的接触图等信息作为模型输入,同样也能提升二面角、二级结构预测性能。
(4)训练样本依然偏少。截至到2022年4月,PDB数据库中通过生物实验手段解析的已知蛋白质结构10.66万条。这些数据无法支持类似BERT大规模模型训练,需要设计对序列特性捕获更好的深度学习模型。
4 结束语
蛋白质骨架二面角是蛋白质结构的重要属性,高精度地预测蛋白质骨架二面角以加速对三维结构构象空间的有效采样,对蛋白质三级结构预测具有重要意义。该文对蛋白质骨架二面角预测算法的发展和领域内最新研究进行了综述,从序列表征、输出、数据集、结构框架等方面介绍算法。同时,对当前二面角预测存在的问题进行了思考。
