Advanced prediction of organic–metal interactions through DFT study and electrochemical displacement approach
2023-03-24AbdelkarimChaouikiWailAlZoubiYoungGunKo
Abdelkarim Chaouiki,Wail Al Zoubi ,Young Gun Ko
Materials Electrochemistry Group,School of Materials Science and Engineering,Yeungnam University,Gyeongsan 38541,South Korea
Abstract Heterocyclic compounds are the promising biological compounds as nature-friendly for the corrosion protection of metallic surface.In this work,three heterocyclic compounds such as 1-azanaphthalene-8-ol (8-AN),2-methylquinoline-8-ol (8-MQ),and 8-quinolinol-5-sulfonic acid (8-QSA) were used as green compounds,and their anti-corrosion performance for AZ31 Mg in saline water was discussed on the basis of impedance interpretation and surface analysis.Findings found that the electrochemical performance was improved in the order of 8-AN> 8-MQ > 8-QSA,demonstrating the electron donor effect of N-heterocycles to form coordination complexes on the magnesium surface.From the electrochemical performance,the protective layer constructed at the optimal concentration reinforces the barrier against aggressive environments,with potential inhibition efficien y of 87.4%,99.0%,and 99.9% for 8-QSA,8-MQ,and 8-AN,respectively.Quantum chemical parameters and electron density distribution for free organic species in the absence and presence of Mg2+ cation were evaluated using density functional theory (DFT).Upon the formation of coordination complexes between organic compound and Mg2+,energy gap underwent change about ΔE=5.7 eV in the 8-AN/Mg2+ system.Furthermore,the adsorption of heterocyclic compounds on Mg surface reveals the formation of strong covalent bonds with Mg atoms,which further confirme by the electron density difference and projected density of states analyses.Based on theoretical calculations,three inhibitors can adsorb on the metal surface in both parallel and perpendicular orientations via C,O and N atoms.In the parallel configuration the C-Mg,N-Mg and O-Mg bond distances are between 2.11 and 2.25 ,whereas the distances in the case of perpendicular adsorption are between 2.20 and 2.40 (covalent bonds via O and N atoms).The results indicated that parallel configuration are energetically more stable,in which the adsorption energies are -4.48 eV (8-AN),-4.28 eV (8-MQ) and -3.82 eV (8-QSA)compared to that of perpendicular adsorption (-3.65,-3.40,and -2.63 eV).As a result,experimental and theoretical studies were in well agreement and confir that the nitrogen and oxygen atoms will be the main adsorption sites.
Keywords: Organic layer;Heterocyclic compounds;Electrochemical behavior;DFT modeling.
1.Introduction
Magnesium and its alloys are attracting more and more attention in the various industries such as aerospace field electronic devices and automobiles industry,etc.,owing to their interesting properties [1–3].This is because the exceptional characteristics of Mg alloy related to its low density and good mechanical properties,i.e.,high strength-to-weight ratio and high thermal conductivity [4,5].Despite these unique properties,Mg alloy suffers from corrosion owing to its high chemical reactivity in the aggressive solution,leading to instability of metal particles that usually low its surface resistance [6–8].Therefore,the formation of an oxide layer on the Mg surface in the presence of saline water offers limited corrosion protection [9,10].
In order to address this issue,different methods such as anodic oxidation,electrodeposition,chemical and physical vapor deposition,conversion coatings and organic coating have been developed to protect the surface of Mg in aggressive solutions [11–18].Investigations on the preparation of these anticorrosion layers must consider the aspects of resource availability and cost-effectiveness for enabling the use of greener approaches and techniques.In this regard,organic corrosion inhibitors (CIs) have drawn tremendous recent attention owing to their effective use as anticorrosion agents in a broad spectrum of corrosive environments [19–21].The use of CIs is a highly effective strategy for the corrosion protection of Mg alloys in aggressive solutions.Furthermore,several types of green CIs,such as plant extracts,oils,fresh and expired drugs,surfactants,amino acids,ionic liquids,and organic compounds,have been developed [22–26].Nevertheless,a survey of the relevant studies provides insights about the considerable challenges involved in developing highly effective CIs for Mg alloys owing to its high chemical activity[27–33].To the best of our knowledge,no attempts to thoroughly clarify the boding mechanism donor-acceptor systems using theoretical studies of heterocyclic aromatic compounds towards AZ31 Mg alloy in aggressive solution has been reported in the literature.Organic CIs are generally used to protect stainless steel against corrosion because they chemical adsorb on the surface acting as a protective coating on anodic and cathodic areas simultaneously.For instance,natural polymers[34],aninoic surfactants[35],organic acids[36],8-hydroxyqunoline [37],amine [28] and sodium lauryl sulfate[38] into Mg surface have reported by several researchers to control the corrosion resistance by means of chemical,electrochemical,and surface analyses.Despite these investigations that have been performed so far to study the growth mechanism and electrochemical performance of CIs on the Mg surface,the working mechanism of CIs is still not understood.In other word,the molecular electronic parameters of free CIs and classical MD simulations cannot provide information about bonding,electron transfer,charge density difference,and density of states,which are of great importance in exploring and a deep understanding of the interaction mechanism CIs and Mg surface.To resolve above-mentioned issues,we reported three class of nitrogen-containing heterocyclic compounds as green and sustainable inhibitors with exceptional corrosion protection to control the corrosion of Mg alloy in aggressive solutions.Moreover,we demonstrate the interfacial mechanism to achieve a complete understanding of the inhibitor-Mg interactions using modern concepts in theoretical chemistry together with experimental evaluations,especially those enabled to elucidate the intrinsic properties of CIs and their adsorption behaviors.
The mechanism was studied using two levels of theory as follows.First,density functional theory (DFT)-based computations were performed to evaluate the reactivities of several organic compounds and capture the ability of complexity with Mg ions during corrosion protection mechanism from a theoretical perspective.DFT calculations assisted in determining the fundamental properties of the organic CIs and their interactions with Mg ions.Second,first-principle DFT calculations were used to explore the interfacial behavior of N-heterocycles compounds on the Mg surface by elucidating the types of intermolecular interactions and adsorption configurations In addition,experimental analyses were performed to confir the relationship between the inhibition performance and the electronic structure calculations,considering the metallic surface.
The present study aimed at determining the corrosion protection ability of three sustainable,organic CIs,namely,1-azanaphthalene-8-ol (8-AN),2-methylquinoline-8-ol (8-MQ),and 8-quinolinol-5-sulfonic acid (8-QSA),for the AZ31 alloy in a 3.5% NaCl solution.Detailed theoretical and experimental insight were obtained on the basis of quantum chemical calculations,electrochemical impedance spectroscopy (EIS),potentiodynamic polarization (PDP),and microstructure analysis.As a proof-of-concept,this study raises important questions regarding the impact of the electronic properties and functional groups of CIs on their adsorption strength on the Mg surface,and consequently,on the corrosion resistance.Furthermore,based on the structural analysis of the CIs and their complexes with Mg2+,the DFT approach was employed to provide additional evidence concerning charge transfer and charge distribution in the ligand–magnesium systems.The adsorption behavior of the investigated molecules were examined based on these results.
2.Theoretical and experimental approaches
2.1.Computational details
2.1.1.Modeling of metal–inhibitor complex
Theoretical calculations were performed using the principles of conceptual density functional theory (DFT) to examine the interactions between Mg2+and the following three CIs derived from quinoline compounds: 8-AN,8-MQ,and 8-QSA.For this purpose,the interactions between Mg2+and molecule complexes were modeled using Gaussian 16 W software [39].The geometries of all the metal-ion–inhibitor systems were optimized using the B3LYP functional with the 6–311++G(d,p) basis set in water by implementing the polarizable continuum model (PCM) model in the Gaussian package[40].Frequency calculations were performed at the same level of theory to ensure that the modelled systems corresponded to a local minimum potential energy.Furthermore,additional electronic structure analyses involving the electron localization function(ELF)[41],density of states(DOS),and electron density difference (EDD) [42] of the studied systems were calculated using the Multiwfn 3.8 code on the optimized systems [43].Finally,quantum chemical (QC) descriptors such as the highest occupied molecular orbital(HOMO)and lowest unoccupied molecular orbital (LUMO) energies,energy gap(Eg),electronegativity (χ),and global hardness (η) were calculated within the framework of molecular orbital theory to predict the chemical reactivities of the inhibitor compounds.These QC parameters have been mathematically described in detail elsewhere within the framework of finit difference approaches [44].
2.1.2.Modeling of adsorbed inhibitors on a Mg (0001)surface
First-principles DFT calculations were performed to explore the adsorption mechanism of the organic CIs on the Mg surface.A Mg (0001) simulated surface was constructed,and a 5×5 supercell slab was created with a vacuum slab of 20in the z-direction using the Materials Studio 6.0 program[45].Systems with and without adsorbed molecules were modeled based on ultrasoft pseudopotentials using the CASTEP package.Calculations were performed using the generalized gradient approximation in the Perdew–Burke–Ernzerhof exchangecorrelation functional (GGA-PBE) [46].For all calculations,the electronic wave functions were expanded using a planewave cut-off energy of 480 eV.Brillouin zone analysis was performed using a Monkhorst–Pack grid using a smearing parameter of 0.03 Ry,and the relaxations were carried out using the Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm with a force convergence cut-off of 0.01 eV/per atom[47].The modern dispersion-corrected method (DFT-D2) is used as recommended by Grimme to explore long-range interactions [48].During optimization,only the bottom two layers were constrained,and all degrees of freedom were relaxed.In this regard,the Brillouin zone was limited to the gamma point owing to the large size of the systems of interest.Furthermore,parallel and perpendicular initial-adsorption modes of the inhibitor molecules on the Mg surface were considered,and the systems were confirme to be converged when all the forces were less than 0.01 eV/.The adsorption energy (Eads) was calculated based on the total energies of the optimized metal–inhibitor (Esurf+inh),Mg surface (Esurf),and isolated inhibitor (Einh) using the following equation:
2.2.Materials and electrolyte
An AZ31 Mg alloy with the following chemical composition(in wt.%)was used in this investigation:2.89 Al,0.96 Zn,0.31 Mn,0.15 Fe,0.12 Si,and balanced Mg is the material used in this investigation.Coupon surfaces with dimensions of 8 mm×8 mm×4 mm were polished with abrasive SiC papers of progressively fine grain sizes (SiC #400 up to SiC #1200),and rinsed sequentially with ethanol and distilled water prior to immersion in the corrosive solution.Corrosion tests with and without inhibitor molecules at different concentrations were conducted in 3.5 wt.% NaCl solution,which was prepared using distilled water,and their inhibition performances were compared.
2.3.Electrochemical studies
Electrochemical techniques such as EIS and PDP were performed in a three-electrode system using a ZAHNER IM6ex electrochemical device (Gamry Interface 1000),in which Ag/AgCl and Pt were used as the reference and counter electrodes,respectively.For the PDP scans,the measurements were conducted from −0.4 V to 0.4 V with respect to opencircuit potential (OCP) at a scan rate of 1 mV/s.EIS tests were performed from 105 Hz to 0.1 Hz at 10 points per decade using AC signals of 10 mV perturbation.All electrochemical measurements were performed in a NaCl solution at ambient temperature and repeated at least three times to ensure the reliability of the data.
The protective abilities of the tested inhibitors in terms of their inhibition performance in both techniques were evaluated using corrosion current density and charge-transfer resistance according to the following equations:
2.4.Characterization techniques
The microstructures of the AZ31 samples were analyzed by examining their surface morphologies using a field emission scanning electron microscopy apparatus (SEM;HITACHI,S-4800).X-ray diffraction(XRD;RIGAKU,D-MAX 2500),in the 2θrange of 30°–90° and a step width of 0.05°,along with X-ray photoelectron spectroscopy (XPS),was employed to determine surface components of the prepared samples.Furthermore,Fourier transform infrared spectroscopy(FT-IR,Perkin Elmer,Spectrum 100) was employed to examine the corrosion products on the AZ31 surface.
3.Results and discussion
3.1.DFT calculations based on charge-transfer properties
3.1.1.Qualitative prediction of inhibition performances based on molecular properties
Functional properties of the selected inhibitors and complexation formation with Mg2+cations were investigated to determine the optimal CI and reduce the cost and time required for experimental analysis.The chemical descriptors and electron properties of both systems,that is,the three isolated molecules (8-AN,8-MQ,and 8-QSA) and molecule–Mg2+complexes,are listed in Supplementary Table 1,and their molecular orbitals (MOs) are depicted in Fig.1a–c.
The results indicate that the 8-AN inhibitor in the isolated form is an acceptable electron donor,but its electron accepting ability is low.Therefore,the reactivity of 8-AN is less than those of 8-MQ and 8-QSA,if the energy gap is assumed to represent the molecular reactivity of organic compounds [49–58].In addition,the MO distribution is primarily related to the functional groups in molecules as donor or acceptor groups that are located on the phenyl ring linked to N and O atoms,except for the LUMO state of 8-AN,which provides a smaller contribution.On the other hand,quantum factors such as chemical hardness and electronegativity were used to examine the chemical reactivity.However,there is no agreement with the experimental results with respect to these parameters.Therefore,the possibilities of designing effective inhibitors and predicting their inhibition performances based only on their QC properties in their isolated states must be thoroughly investigated.
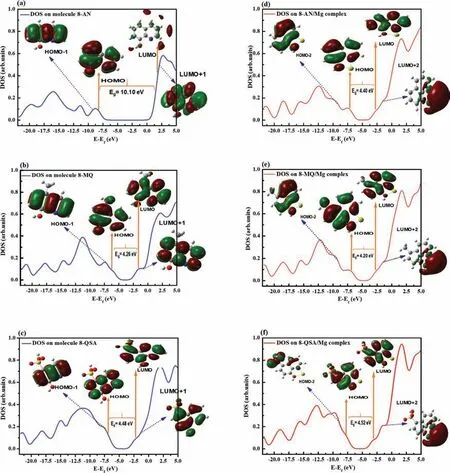
Fig.1.(a–c) DOS plots of the studied inhibitors and (d–f) their complexes with Mg2+ in water,along with their molecular orbitals.
Most DFT-based studies on corrosion inhibition have focused on QC parameters,which may not always be reliable for clarifying inhibition performance trends and describing inhibitor–metal interactions.Therefore,additional investigations that considered different variables (solvent effect,presence of ions,and metal surface) were undertaken prior to establish an association between the QC descriptors and adsorption efficien y.Considering the MO properties of the aforementioned three molecules,a detailed interaction mechanism can be proposed by addressing the roles of donor and acceptor groups during the interactions between the 8-AN,8-MQ,and 8-QSA inhibitors and Mg2+cations.The QC parameters can presumably be correlated well with the proposed model,in which the changes in electronic and charge-transfer properties during the formation of ligand–magnesium complexes are probed.Therefore,appropriate DFT-based computational methods are required to assess the effects of charge transfer on the QC parameters due to complex formation.
3.1.2.Computational perspectives on the considerable interactions between Mg2+ and inhibitor molecules
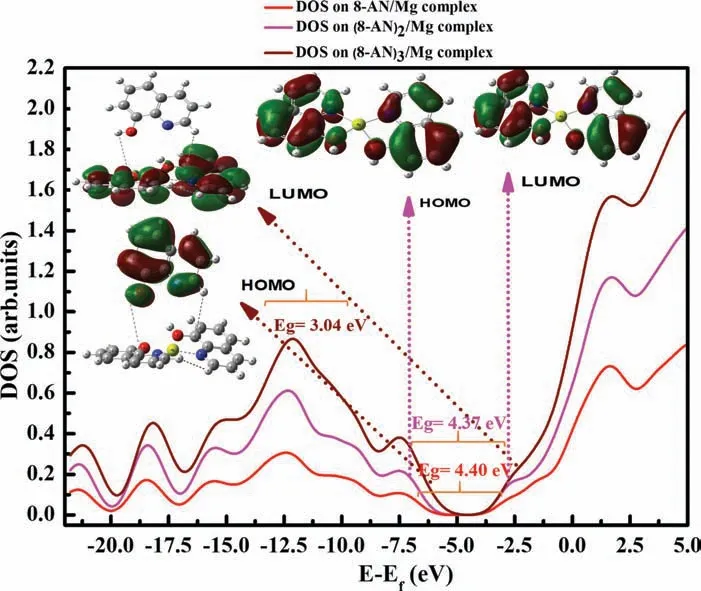
Fig.2.Calculated DOS plots of the Mg(8-AN),Mg(8-AN)2 and Mg(8-AN)3 complexes along with their molecular orbitals.
The abilities of three inhibitors to form complexes with Mg2+were investigated to gain insights into the effects of molecule–Mg2+interactions on the overall electronic properties of the complex systems.Therefore,the physical or chemical coordination of the inhibitors to form complexes with Mg2+was evaluated based on the DOS,ELF,EDD,enthalpy change (ΔH),and energy (ΔG,Einteration) analyses.The DOS of 8-AN,8-MQ,and 8-QSA were computed after complex formation,and the obtained results,along with their MOs,are shown in Fig.1d–f.The DOS was simulated based on the distribution of MO energy levels,and the electron properties of the organic compounds were found to undergo several changes after complex formation.For example,during the formation of the AN-8/Mg complex,the LUMO of 8-AN is distributed around the entire molecule.Moreover,certain changes are visible in both parts of the DOS diagrams after complex formation;the considerably larger DOS compared to that prior to the interactions suggests the existence of certain important orbital interactions between the molecules and Mg2+cations.Interestingly,the concept of DOS is useful in this case to elucidate the changes in the energy gap after complexation.Interestingly,the energy gap is significantl reduced from 10.10 eV to 4.4 eV during the interaction between 8-AN and Mg2+,indicating that the changes in charge density occur upon interaction,resulting in straightforward charge transfer in this molecule.This is possibly due to the important role played by the electron-donor group,which assists in establishing coordination bonds and facilitating charge transfer.Based on these results,the proposed calculation model was confirme to provide reasonably consistent evidence of an association between molecular reactivity during molecule/Mg interactions and the experimental results.Consequently,8-AN exhibits remarkable reactivity with respect to its intra-and inter-molecular bonding with Mg2+cations during their interactions.In addition,investigating the effects of the number of 8-AN molecules interacting with Mg2+on the energy gap can provide interesting theoretical insights.The energy gap and DOS spectra of the (8-AN)2/Mg and (8-AN)3/Mg complexes,along with their MOs,are depicted in Fig.2.An increase in the number of organic inhibitors interacting with Mg2+results in a decrease in the energy gap.This can be attributed to the formation of a greater number of chemical bonds between the molecules and Mg2+,resulting in the formation of strong interactions.Therefore,the interaction energies and thermodynamic parameters of all three compounds interacting with Mg2+were calculated to confir these mutual interactions(Table S1).The 8-AN complex has an energy with the most negative value compared to those of the other complexes,indicating that 8-AN forms a highly stable complex with Mg.Furthermore,the higher Gibbs free energy and enthalpy of the 8-AN/Mg complex indicate its superior reactivity for interacting with the Mg cation.
Moreover,a comparison of the electron densities of all three systems before and after complexation in terms of ELF and EDD is presented in Fig.3.The ELF images suggest that the degree of electron localization is high in the 8-AN and 8-MQ compounds compared to that of 8-QSA.In addition,the molecular structures change,and N and O atoms form bonds with Mg after the complex formation,which affects electron transfer and consequently induces changes in the energy gap,as discussed earlier.The EDD maps,as shown in Fig.3e–g,are highly beneficia for identifying and understanding the changes in electronic behavior due to electron localization.The aqua and dark blue isosurfaces represent regions of increased and decreased electron densities after the coordination of the organic compounds,respectively.The fact that electron density is the highest around the Mg atom is particularly noteworthy.In addition,the electron density distribution with respect to 8-AN remarkably increases the coordination bonds between the N and O atoms and Mg.
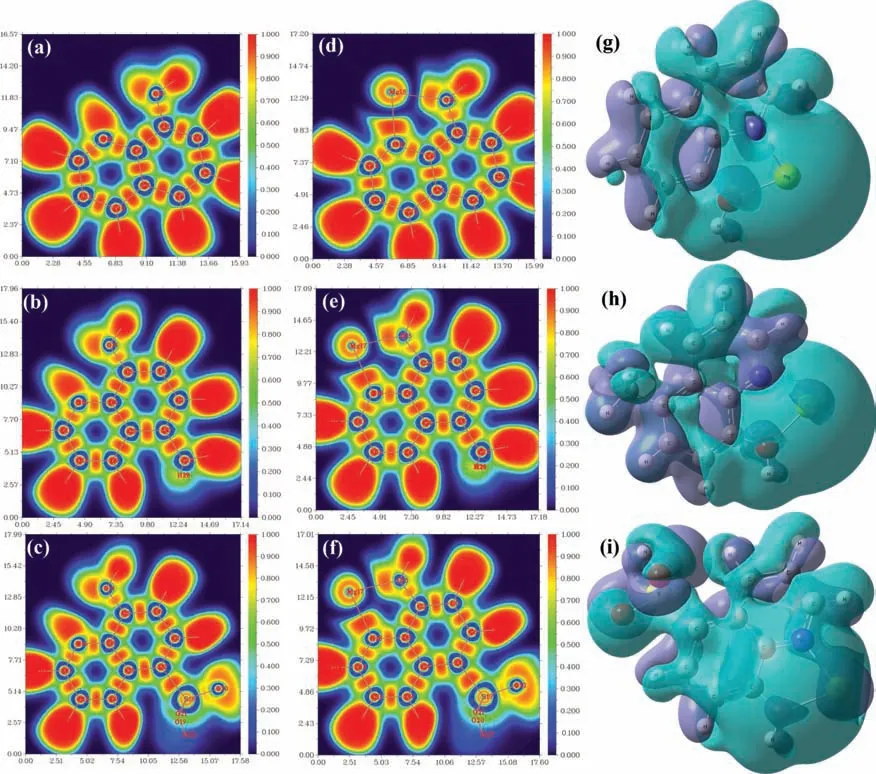
Fig.3.ELF plots of molecules (a,b,c) before and after interactions (d,e,f) with Mg2+.(g,h,i) Isosurface EDD maps of the Mg/inhibitor complexes.
These results suggest that the interaction mechanism is more stable in the presence of an electron-donating group and that hydrogen can readily interact with the Mg cation [59,60]compared to sulfonic acid,which has an electron-withdrawing effect.
3.2.DFT modeling of the interaction modes with respect to the Mg(0001) surface
3.2.1.Parallel and perpendicular adsorption configu ations
First-priciples DFT simulations were performed to gain detailed insight into the actual inhibition mechanism and to validate the structural geometries of the CIs on the surface of Mg,which can possibly affect the adsorption mechanism.The intermolecular interactions of 8-AN,8-MQ,and 8-QSA were modeled by selecting different initial distances between the reactive sites and the surface.Figs.4 and 5 illustrate the interactions between the parallel and perpendicular geometries of the inhibitor compounds before and after adsorption on the Mg surface,respectively.Fig.4 indicates that all three organic inhibitors are adsorbed in parallel on the Mg surface in a similar manner.The results clearly indicate that the N and O atoms are bonded to the adjacent Mg atoms with bond lengths between 2.11(dN−Mg) and 2.32(dO−Mg),which are close to the sum of the covalent radii of N,O,and Mg (rN+rMg=0.71+1.41=2.12,rO+rMg=0.66+1.41=2.07).In addition,8-AN and 8-HQ bind to Mg atoms in their parallel states via the C2atoms and C1atom of the aromatic ring with bond lengths of 2.36and 2.42,respectively.Therefore,N,O,and C atoms,which facilitate the formation of chemical bonds with the Mg surface,are involved in the adsorption-induced interaction mechanism.To further examine the adsorption strengths of these molecules in terms of their bonding tendencies with the Mg surface,the adsorption energies (Eads) of all the systems were estimated.A comparison of the Eadsvalues,as shown in Supplementary Table 2,indicates that strong adsorption of molecules on the metallic surface occurs in the following order: 8-AN>8-MQ>8-QSA.Therefore,the adsorption of molecules is presumably affected by the electron-donating groups in conjunction with the reactive sites,which leads to strong bonding and superior stability of the molecules.However,the QSA/Mg adsorption system containing the–SO3H functional group appears to have a low adsorption affinit,possibly because the sulfonic acid group,owing to its electron-withdrawing effect,significantl diminishes the electron density of the aromatic ring,thereby causing the low reactivity of the hydroxyl group and donor sites during interaction process.
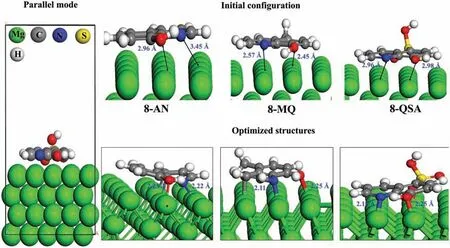
Fig.4.Top plots: Initial and fina adsorption geometries of the studied inhibitors on the Mg (0001) surface in the parallel mode.Bottom plots: EDD plots corresponding to the preferred adsorption orientations.(For interpretation of the references to colour in this figur legend,the reader is referred to the web version of this article.)
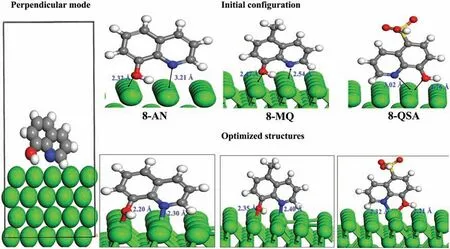
Fig.5.Top plots: Initial and optimized adsorption geometries of the inhibitors on the Mg (0001) surface in the perpendicular mode.Bottom plots: EDD plots corresponding to the preferred adsorption configurations (For interpretation of the references to colour in this figur legend,the reader is referred to the web version of this article.)
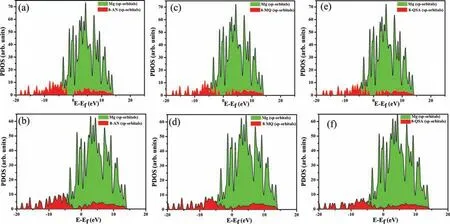
Fig.6.PDOS analysis of molecules before (a,c,and e) and after (b,d,and f) adsorption on the Mg (0001) surface,and that of the Mg atoms beneath them.
The perpendicular adsorption behavior was also investigated using the DFT-D2 model.As shown by the adsorption geometries of the molecules in Fig.5,the inhibitor compounds interact with the Mg surface via N and O atoms to form chemical bonds.This is confirme by the bond lengths of N–Mg and O–Mg,which decrease compared to their initial lengths.In addition,the noteworthy geometrical change due to the vertical adsorption is presumably because of the tendency of the molecules to exhibit a tilted orientation,which explains the absence of Mg–C bonds.Furthermore,the estimated Eadsvalues reveal that all three compounds are less likely to adsorb on the Mg surface in the vertical adsorption mode than in the parallel mode.Moreover,compared to 8-QSA,8-AN and 8-MQ are more likely to be adsorbed on the Mg surface owing to their superior electron-rich nature.The optimized bond lengths (N–Mg and O–Mg) considerably decrease in the parallel mode,and their chemisorption is stronger than that in vertical adsorption.Therefore,the inhibitor molecules,particularly 8-AN,maximize the contact area in their parallel geometries.
These results show adequate agreement with theoretically obtained results on the adsorption of 8-HQ and its derivatives on an Al(111) surface [61].Additionally,these finding strongly support the enhanced adsorption efficien y of 8-AN compared to those of 8-MQ and 8-QSA.Consequently,these results provide solid evidence from a theoretical perspective and can successfully explain the experimentally obtained inhibition trends.Furthermore,examining charge transfer relative to the CIs and the Mg surface could provide more definit ve evidence for comprehensive understanding of the adsorption processes,which will be discussed later in this paper.
3.2.2.Projected electron density difference (EDD) of the systems
The concept of EDD was used effectively in this study to capture the formation of covalent bonds during the interactions and to provide valuable information regarding charge displacement in the electronic structures of molecules adsorbed on the Mg surface.Because of the binding of molecules to the Mg surface,changes in electron distribution were calculated via EDD by subtracting the electron density variation of isolated molecules (ρmol) and that of the Mg(0001) surface (ρsurf) from the global electron density of the Mg–inhibitor systems (ρsurf/mol) using the following correlation:
The difference maps of electron density represented in Figs.4 and 5 with an isosurface of ±0.002 e/3reveal the presence of a charge density between the O,N,and Mg atoms,which also suggests the formation of covalent bonds for both configurations The blue and dark golden regions represent charge accumulation and charge loss,respectively.Changes in the EDD were expected to occur in electron-donating groups and exhibit a bias toward the Mg surface.Interestingly,the charge accumulation is more significan in the parallel configuration and is localized in the Mg–N,Mg–O,and Mg–C bonds and over the aromatic rings.These results confir the strong interactions between the inhibitor compounds in the parallel mode and the Mg surface.However,the charge redistribution in the vertical adsorption scenario is mainly shared between N,O,and Mg atoms;therefore,the molecules exhibit weaker interactions with Mg atoms compared to those in the parallel geometries,which further confirm the adsorption energies of the studied systems.
In addition,the charge accumulation in the case of 8-QSA was found to be negatively affected by the electronwithdrawing group (SO3H) based on the isosurfaces;this diminishes the charge transfer from the molecule to the surface,leading to the charge redistribution being biased toward the N and O atoms.
3.2.3.Projected density of state (PDOS)
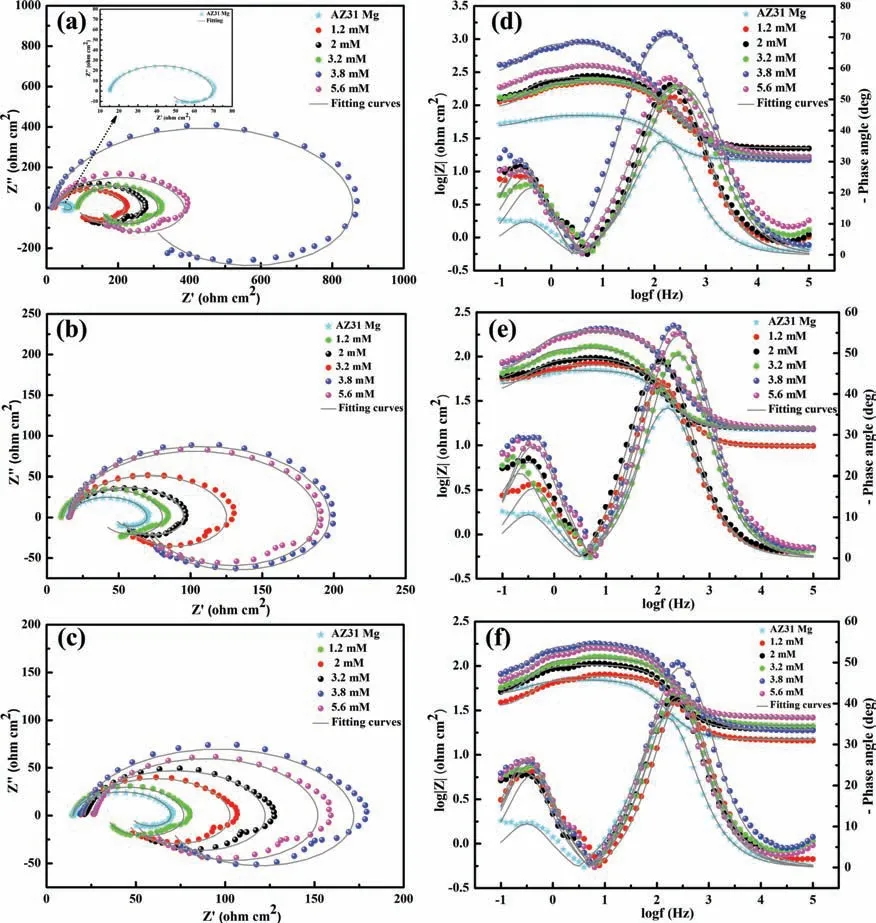
Fig.7.(a-c) Nyquist and (d-f) Bode plots obtained by immersing AZ31 in a 3.5 wt.% NaCl aqueous solution without and with different concentrations of inhibitors at room temperature.
The molecule–surface bonding and the contribution of the elements to the overall interactions at the electronic level during adsorption were examined by projected density of states(PDOS) analysis.Fig.6a-f summarizes the PDOS for the reactive centers of molecules located 7above the Mg (0001)surface before and after adsorption in the most stable config urations.The Mg-sp bonds contain several molecular states of sp-orbitals prior to the interactions (Fig 7a,c,e),suggesting the suitability of these orbitals for hybridization with sporbitals of Mg atoms during adsorption.The interaction of the CI molecules with Mg via C,O,and N atoms,that is,adsorption,results in broadening of the peaks and slight decreases in intensities compared to the sharper,intense peaks prior to adsorption.These results indicate strong hybridization between molecular orbitals and Mg-sp orbitals.Therefore,the hybridization,and consequently,the interactions via the bonded atoms in the case of the 8-AN/Mg system,are more significant This is because C,O,and N atoms have larger contribution of lone pair to the band formation,which lead to the charge transfer,enabling a significan effect compared to that in the other molecules.These observations strongly support the inhibition performance behavior.
3.3.Electrochemical displacement
3.3.1.Compounds containing donor and acceptor groups
The effects of concentrations of 8-AN,8-MQ,and 8-QSA on the characteristics of the anticorrosion barrier formed on the AZ31 Mg substrate were subsequently investigated(Fig.7a-f).Overall,the Nyquist and Bode diagrams indicate that the inhibition effect is significantl enhanced with increasing amount of inhibitors up to an optimal concentration of 3.8 mM.The impedance responses for all systems and concentrations primarily consist of one capacitive loop and one inductive loop at high and low frequencies,respectively.Interestingly,the results corresponding to the bare AZ31 Mg alloy without inhibitors are similar to those obtained previously [62–65]. Fig.7a-c suggests that all the systems with inhibitors exhibit similar behavior,indicating the occurrence of a similar corrosion inhibition effect.Larger capacitive loops are obtained at the optimal concentration,implying that the systems exhibit superior performance against corrosion of the Mg substrate.In addition,the optimization of the inhibitor content and its effects on the AZ31 Mg surface reveal that the best inhibition performance was achieved with the addition of 8-AN,as indicated by the larger diameters of the capacitance loops.Therefore,among all the studied inhibitors,the AZ31 Mg surface is presumably covered to a greater extent by adsorbed layers of 8-AN (Fig.7a) when displacement reactions occur between metal and inhibitor,which enables a more effi cient blocking of charge and mass transfer for mitigating the corrosion of metal alloys.
Furthermore,the corrosion resistances of the different systems were compared using the values of the impedance modulus,Zmod,as indicated in the Bode diagrams.Typically,a larger Zmodimplies a better anticorrosive performance of the inhibitor fil [66].The Bode diagrams indicate that the Zmodof the barrier fil of 8-AN is approximately 103Ω•cm2at low frequencies (Fig.7d),which is higher than that of the AZ31 substrate (101.5Ω•cm2).In addition,these results show that the phase angles of all the systems exhibit two peaks,which are related to two time constants,the firs of which appears in the intermediate-frequency region;this is possibly related to the electric double-layer capacitance and charge-transfer response at the AZ31 Mg/electrolyte interface(change in the phase angle from −40° to −75°).In this case,the barrier layer primarily consists of Mg(OH)2as a corrosion product from the dissolution of Mg that occurred during the cathodic reaction.The second time-constant corresponds to an inductive loop that can presumably be attributed to the high concentration of Mg2+during an intermediate step in the process of corrosion.These results demonstrate that the protective layer formed owing to adsorption of the 8-AN inhibitor on the AZ31 Mg alloy provides superior anticorrosion protection,and can significantl reinforce the barrier against dissolution;this is mainly because of the noteworthy electronic properties of 8-AN relative to those of the other compounds.
Quantitative analysis based on the impedance spectra was subsequently conducted to gain insight into the corrosion inhibition characteristics of the different systems.To this end,an equivalent circuit (EC) model represented by Rs(QdlRct(RLL))was employed (Fig 9b).This EC model quantifie all electrochemical parameters in the absence and presence of inhibitors by considering the physical and chemical features of electrochemical processes.In the EC model,Rsis the electrolyte resistance,RLis the resistance related to the inductance (L),and Qdland Rctare the constant phase element and the charge-transfer resistance at the Mg interface,respectively.Supplementary Table 3 provides all parameter values obtained from the EC model,which reveals that the fil resistance increases with increasing inhibitor concentration.The protective layer constructed at the optimal concentration (3.8 mM) reinforces the barrier against corrosion attacks,with potential inhibition performances of 93.5%,69.8%,and 64.2% for 8-AN,8-MQ,and 8-QSA,respectively.On the other hand,the Qdl(Ydl) values decrease with increasing concentrations of the three inhibitors,suggesting that the most homogenous steel surface was obtained at a concentration of 3.8 mM of inhibitors;however,no significan changes were observed in terms of the Rsvalues.Interestingly,the protective layer deteriorates if a considerably high amount of the inhibitor is added to the solution,as demonstrated by the low Rctvalues.Consequently,the optimal amount of the inhibitor must be determined to ensure ideal protection against corrosion.Based on the aforementioned results,effective protection can be achieved using 8-AN owing to its functional properties.
In addition to the EIS studies,PDP experiments were conducted to provide more information regarding the corrosion behavior of AZ31 Mg in 3.5 wt.% NaCl at room temperature.The corresponding PDP curves for all three systems are shown in Fig.8a-c,and the derived electrochemical parameters are listed in Supplementary Table 4.
The PDP curves suggest that the presence of inhibitor compounds results in substantial changes in terms of cathodic and anodic current densities,indicating that the addition of the quinolinole derivatives hinders the corrosion attacks compared to the uninhibited solution by means of a passive fil on the AZ31 Mg surface.A significan difference in corrosion current density can be observed in the case of 3.8 mM 8-AN (28.7×10−9A/cm2) compared to those of bare AZ31 (192.0×10−6A/cm2) and AZ31 with the addition of 8-MQ (1.79×10−6A/cm2) and 8-QSA (23.4×10−6A/cm2) at similar concentrations.As mentioned earlier in the EIS analysis,an increase in the inhibitor amount beyond 3.8 mM leads to variations in the anticorrosion ability of the protective film Additionally,Ecorrshifts in a positive direction,except that of the AZ31 Mg/8-MQ system,which slightly shifts to negative values;this indicates an improvement in the polarization resistance of the protective layer on the metal surface.Moreover,relative to the uninhibited system,the maximum shift of Ecorris lower than 85 mV,representing the mixed inhibition behavior.Furthermore,the variations inβcandβaafter the addition of inhibitors demonstrate the effects of the studied compounds on both the kinetics of hydrogen evolution and anodic dissolution of the AZ31 Mg alloy.A feasible explanation involves the inhibition possibly occurring mostly by the blocking of active corrosion sites on the metal surface because of the adsorption of the CIs.
Notably,the adsorption abilities of the three investigated compounds in terms of protection efficien y adhere to the following order: 8-AN>8-MQ>8-QSA.Moreover,this trend has been elucidated on the basis of the electronic molecular properties of the inhibitors.Therefore,the 8-AN compound was confirme to cover a considerable surface area on the AZ31 Mg alloy,which further strengthens the results obtained by EIS.Finally,EIS was employed again to analyze the effects of consecutive immersion time on the electronic molecular properties,particularly the electron-donating ability,of the investigated molecule.

Fig.8.Polarization curves of the AZ31 Mg alloy in a 3.5 wt.% NaCl solution without and with different concentrations of (a) 8-AN,(b) 8-MQ,and (c)8-QSA,along with the (d) FT-IR spectra recorded at an inhibitor concentration of 3.8 mM.
3.3.2.Influenc of immersion time on behavior of the protective layer
In addition to the effects of concentration,the stability and durability of the anti-corrosion layer formed during corrosion inhibition of the AZ31 Mg alloy are essential factors that should be considered.Therefore,the inhibition behavior of 8-AN was investigated in terms of long-term immersion(1,2,6,12,24,72,and 120 h) to determine the stability of the corrosive performance of this inhibitor.Fig.9a shows the Nyquist diagrams of the protective layer containing the optimal concentration of 8-AN obtained using different immersion times;the corresponding fitte data are listed in Supplementary Table 5.
The Nyquist plots indicate that the mitigation of the corrosion of AZ31 Mg is significantl affected by the duration of immersion;moreover,the formation of the stable protective region follows a similar behavior for different time intervals,as determined by the inductance and capacitance loops.The larger dimensions of the capacitance loops suggest the positive impact of 8-AN on the corrosive resistance over time,except for the 12 h immersion scenario,in which the corrosion resistance decreases compared to that of the 6 h immersion scenario.Interestingly,even after 120 h of immersion,the AZ31 Mg/8-AN system continues to exhibit a superior anticorrosion performance,which can be attributed to the strong adsorption of the inhibitor.This confirm the adsorption and accumulation of large amounts of 8-AN on the AZ31 surface with prolonged immersion periods;therefore,the protective layer becomes more stable and protective.Moreover,a longer duration is required for the inhibitor to inhibit the Mg surface corrosion.A more conclusive comparison is presented in terms of Rctfor various immersion times,and the other parameters calculated using the EC model are not discussed here owing to similarities in the features to those mentioned in the aforementioned section.Briefl,the AZ31 Mg/8-AN system exhibits an improvement in Rctat prolonged immersion periods,indicating that the protective fil formed by 8-AN is not easily ruptured.These results demonstrate the potential of 8-AN as a candidate for improving the durability of anti-corrosion layers,which is due to its high electron-donating character compared to those of the other tested compounds,as revealed by the theoretical calculations.
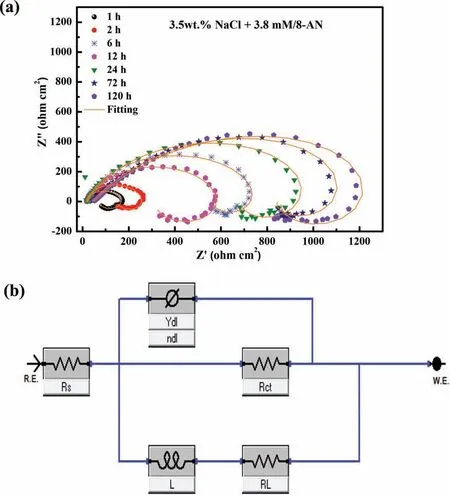
Fig.9.(a) Time-dependent impedance responses for Mg AZ31 samples after immersion in NaCl solution containing 3.8 mM 8-AN and (b) equivalent circuit model used to interpret the impedance responses.
Morphological analysis of the AZ31 Mg/8-AN system was subsequently conducted to further correlate the electrochemical results and characterize the effects of both immersion time and concentration of 8-AN on the microstructure of AZ31 in a corrosive solution.
3.4.Microstructural analysis of protective layer
SEM images of AZ31 Mg substrates immersed in different concentrations of 8-AN (1.2,3.2,3.8,and 5.6 mM)were obtained to characterize the microstructure evolution of the organic layer;coatings with fl wer-like structures were gradually developed on the substrates,as shown in Fig.10a–f.
The prepared coating clearly becomes more compact and offers greater protection with an increase in the concentration to 3.8 mM,which successfully enhances the barrier property of the obtained organic layer.The organic layer (Mg(8-AN))obtained during immersion in a solution containing 8-AN was simultaneously analyzed by XRD to further identify the products on the metallic surface.Fig.10g,h shows that the main peaks of the organic complexes are consistent with those of previously reported hydroxyquinoline derivative powders[67–69],indicating that fl wer-organic like is mostly composed of Mg(8-AN) compound.The growth mechanism of the Mg(8-AN) organic layer is clarifie by the fact that during the chemical conversion of the metallic surface,the AZ31 alloy releases Mg2+ions by chemical corrosion in neutral media,and the dissolved Mg2+ions can directly bond with organic anions to form stable organic complexes (Mg(8-AN)).
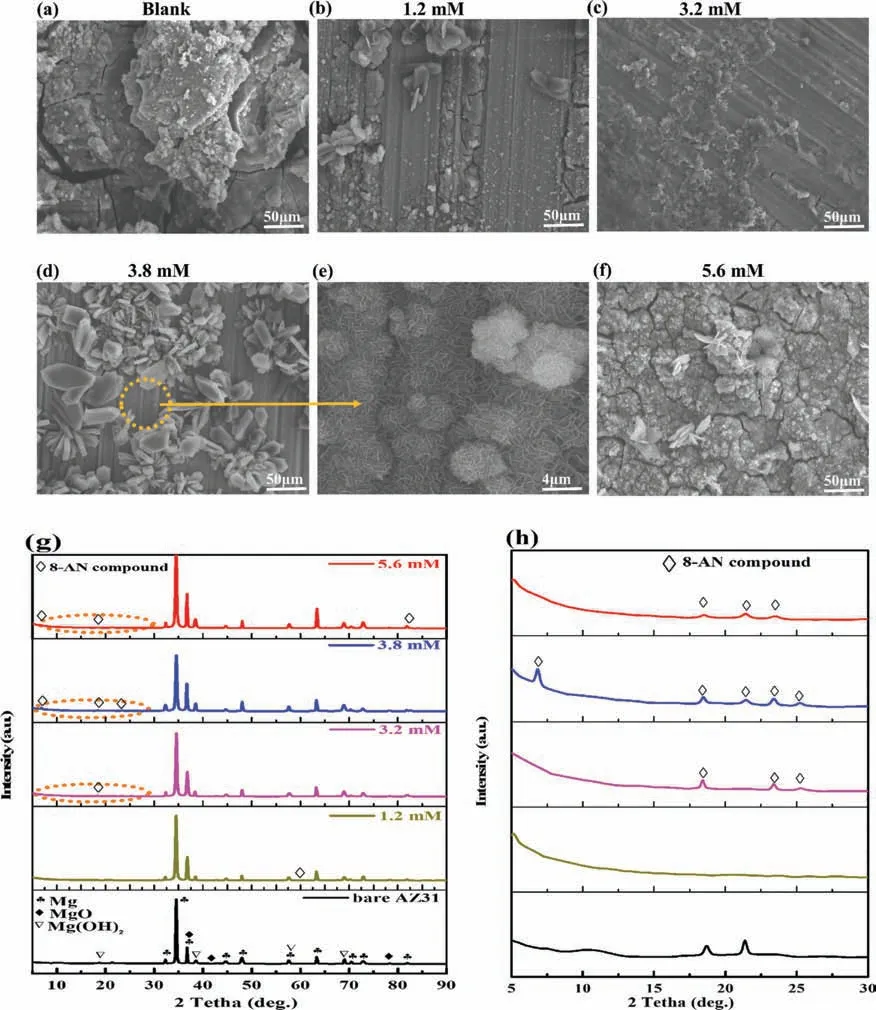
Fig.10.(a–f) Microstructure images of AZ31 immersed in 3.5 wt.% NaCl solution before and after the addition of different concentrations of 8-AN,(e) a high-magnificatio image of (d),(g) XRD patterns obtained in 3.5 wt.% NaCl solution after immersion without and with different concentrations of 8-AN,(h) magnifie values obtained in the 2θ range of 5°–30°.
The resulting organic layer of the AZ31 alloy substrate was analyzed further to identify the chemical structures of the chemical conversion.Fig.11a shows the XRD spectra of the organic layer immersed in 8-AN for different times (1,2,6,12,72,and 120 h).The bands at 18.5° are attributed to the presence of Mg(8-AN)on the organic layer[70].The intensity ratio of (I(Mg(AN)(120h)) reaches ∼1,which suggests that the ratio increases with increasing immersion time in the 8-AN solution up to 120 h.Therefore,the observed differences between the original 8-AN powders and the organic layer indicate that the 8-AN anions chelate with Mg2+in solution,creating a stable Mg(8-AN) phase on AZ31 Mg.
Heterocyclic compounds were selected as the loading organic compounds on the surface of Mg to study the formation of organic layer metals.FT-IR and XPS profile of a plate prepared using the organic compound–Mg were collected to confir the formation of coordination complexes on the Mg surface.The peak shown at 1423 cm−1in FT-IR spectra(Fig.8d) is caused by the stretching of Mg–O bonds,whereas that at 1200–1000 cm−1is attributed to the coordination of S–O and C–O in the ring.Compared with the spectrum of the blank,the primary absorption peaks at ∼3500–3200 cm−1confir the formation of Mg(OH)2on the metallic surface because of immersion in the aggressive solution.The XPS profile (Fig.11b–f) confir the presence of Mg 1s,O 1s,C 1s,and N 1s on the AZ31 Mg surface.Therefore,the N 1s spectra were deconvoluted into two binding energy peaks at 399.2 eV and 398.7 eV,corresponding to the electronic states of Mg–N and C–N,respectively.As shown in Fig.11e,the three fitte peaks in the high-resolution C 1s spectra between 283 eV and 286 eV can be attributed to bonded carbon in the aromatic rings.These results confir the presence of metal oxide,carbon,and nitrogen in the formed organic layer,which supports the FT-IR analysis.
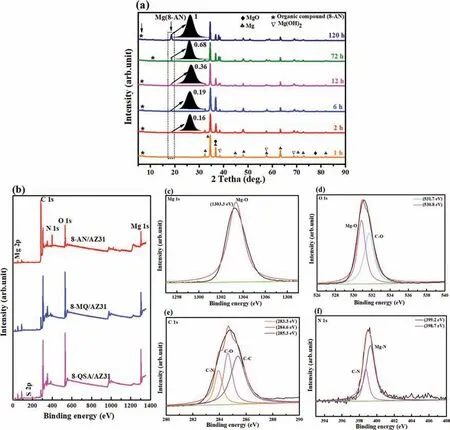
Fig.11.(a) XRD patterns of the organic layer in 3.8 mM 8-AN for various durations,(b) wide XPS profile of the Mg/8-AN system and its high-resolution scans featuring (c) Mg 1s,(d) O 1s,(e) C 1s,and (f) N 1s.
3.5.Comparison with other tested inhibitors for Mg alloy in 3.5 wt.% NaCl solution
To further confir the exceptional corrosion protection of N-heterocyclic inhibitors,8-AN and 8-QM compounds exhibited the lowest value oficorrwhen compared to other reports,such as sodium alginate [35],hydroxylethyl cellulose [35],sodium dodecyl sulfate [71],sodium alginate [72],sodium phosphate [72],sodium alginate [73],triethanolamine [74],and anionic surfactant [75],as shown in Fig.12.These indicated that the addition of 8-AN compound as green inhibitor for AZ31 Mg alloy surface exhibited remarkable corrosion protection at low concentration.This findin would be explained by the chemical nature of N-heterocyclic compounds in which the charge transfer and electron-donor acceptor ability give the opportunities for strong adsorption to form excellent protective barrier for magnesium-based alloys [76].
4.Conclusions
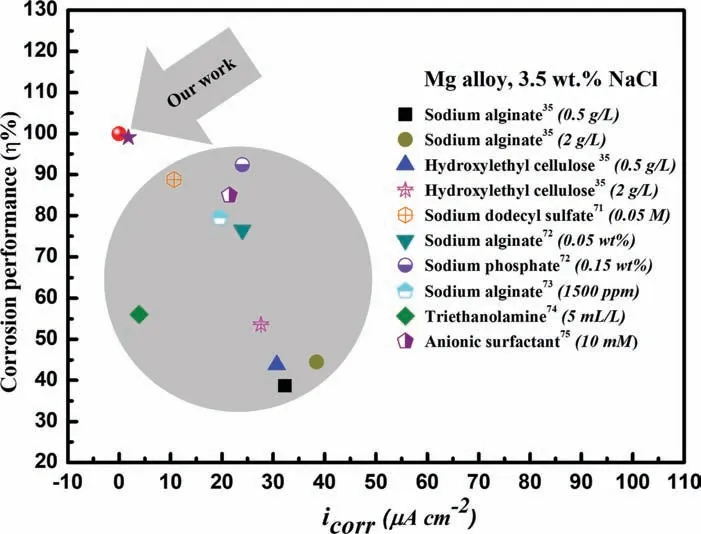
Fig.12.Comparison of icorr and inhibition performance values of the Mg alloy samples treated by several organic inhibitors with different concentrations.For all results,the electrolyte used was 3.5 wt% NaCl solution.
The anticorrosion performance of N-heterocyclic compounds was determined to be superior compared to those of other corrosion inhibitors owing to their electron sites.The coordination between N-heterocycles and Mg ions was successfully established on the metallic surface via chemical bindings for efficien corrosion protection of AZ31 Mg.The electrochemical experiments showed that the adsorption of Nheterocycles modifie the Mg surface and facilitated the formation of 8-AN/Mg formation compared to that of 8-MQ/Mg and 8-QSA/Mg;8-AN reacted with either Mg(II) ions or the metal to form an insoluble and homogeneous coordination complex in aggressive solutions over the long term.The EIS results showed an increase in the corrosion resistance and a decrease in the double-layer capacitance in the presence of the N-heterocyclic compounds.The inhibition ability of the three heterocyclic compounds followed the order 8-AN>8-MQ>8-QSA,which was confirme by surface analyses based on SEM,electrochemical measurements,and theoretical calculations.The formation of strong coordination bonds between the N-heterocycles on the Mg surface was observed in the parallel adsorption mode through the formation of several covalent bonds.Interestingly,DFT-based computational observations revealed that the magnitudes of adsorption energy of the parallel and perpendicular configuration were in agreement with the experimentally obtained adsorption performances.
Declaration of Competing Interest“The authors declare that they have no known competing financia interests or personal relationships that could have appeared to influenc the work reported in this paper”.
Acknowledgments
This work is financiall supported by the National Research Laboratory Project of the National Research Foundation funded by the Ministry of Science and ICT,Republic of Korea (NRF-2020R1A2C2004192).Y.G.K.acknowledges G.Y.H.for research support via the YGY Project (YGY-20150627000).This work is supported by National Research Foundation (NRF) of South Korea (2022R1A2C1004392).
Author contribution statementAbdelkarim Chaouiki: Conceptualization,Methodology,Investigation,Formal analysis,Writing -original draft,Review &editing.Wail Al Zoubi: Formal analysis,Writing -original draft.Young Gun Ko: Supervision,Funding acquisition,Visualization.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.proci.2018.05.113.
杂志排行

Journal of Magnesium and Alloys的其它文章
- Development of high-strength magnesium alloys with excellent ignition-proof performance based on the oxidation and ignition mechanisms: A review
- Development and application of magnesium alloy parts for automotive OEMs: A review
- Recent advances in surface endothelialization of the magnesium alloy stent materials
- Recent developments in high-pressure die-cast magnesium alloys for automotive and future applications
- Exploring the contribution of oxygen reduction reaction to Mg corrosion by modeling assisted local analysis
- Microstructures,mechanical properties,corrosion,and biocompatibility of extruded Mg-Zr-Sr-Ho alloys for biodegradable implant applications