纳米Ce0.92M0.08O2(M=Co,Mn,Fe,La)固溶体的制备及其氧化还原性能
2023-03-22陆旻琳张国芳许剑轶
束 俊,陆旻琳,张国芳,许剑轶
(内蒙古科技大学 材料与冶金学院,内蒙古 包头 014010)
氧化铈具有储量丰富、易开发、性能优良等优点而得到广泛应用,特别是其独特的萤石立方结构及Ce离子的易变价特性,使其能够作为催化剂或助催化剂而广泛应用于多个领域中[1]。二氧化铈作为催化剂的关键特征主要源于由Ce4+还原为Ce3+而引起其晶格内部氧空位的浓度变化。然而,纯CeO2由于热稳定性差和催化活性低,其性能通常不能满足需求。为进一步提高其氧化还原性能,一方面采用纳米化技术来提高比表面积,进而增强其反应活性;另一方面通常利用掺杂离子来优化晶格微观结构,提高氧空位浓度,从而改善其实用性能[2]。通常,掺杂离子自身的化学特性也会对二氧化铈的结构、光谱特征及催化性能产生重要影响。掺杂能够提高CeO2催化性能的一个重要原因在于掺杂离子的尺寸及电子结构。在CeO2晶格中,掺杂离子与氧离子更加倾向于以其自身的氧化物配位,而不是Ce4+的立方配位[3],因此,具有不同电子结构的掺杂离子与氧离子发生更弱或更低的配位,进而表现出比纯CeO2更强的反应活性。
尽管目前关于掺杂CeO2的报道很多[4],但因采用不同的合成方法、不同的掺杂比例及测试条件,导致无法系统分析掺杂离子自身特性对CeO2性能的影响规律。过渡金属离子Co,Mn,Fe具有特殊的电子结构,通常能够表现出多种价态,稀土元素离子La3+具有比Ce离子更加稳定的电子结构及价态。由于这几种离子表现出不同的离子半径、价态及氧化还原特性,因此对二氧化铈的修饰作用也不同。研究表明,Co离子掺杂能够有效提高CeO2基固溶体的催化性能[5],其固溶限>10%,但也有研究证明其固溶限低于6%[6]; Fe离子掺杂CeO2能够有效增大纳米材料的比表面积,提高晶格内的氧空位浓度[7],水热条件下其固溶限为15%[8],Mn离子掺杂可表现出良好的氧化还原性,其固溶限约为20%[9],而同为稀土的La3+掺杂可有效修饰晶格,且固溶限超过20%[10]。
本工作采用水热法制备掺杂浓度均为8%的Co,Mn,Fe,La掺杂纳米CeO2基固溶体,系统研究固溶体的微观结构、光谱特征及氧化还原特性等变化规律,以期探索掺杂离子特性对CeO2结构及性能的影响机制。为进一步研究掺杂离子特性对CeO2微观晶体结构及催化性能的影响机制,系统研究所得固溶体掺杂前后微观结构的变化,通过紫外光谱、荧光光谱、拉曼图谱等手段对纳米Ce0.92M0.08O2(M=Co, Mn, Fe, La)材料的光谱特征进行系统表征,利用化学吸附(TPR)测试掺杂样品的氧化还原特征,从而获得掺杂离子化学特性对CeO2的微观结构、光谱特征及催化性能的影响规律。
1 实验材料与方法
1.1 样品的制备
选用的化学试剂有Ce(NO3)3·6H2O(AR),La(NO3)3·6H2O(AR),Fe(NO3)3·9H2O(AR),Co(NO3)2·6H2O(AR),Mn(NO3)2·4H2O(AR)和NaOH(AR),药品均来自阿拉丁试剂有限公司。
先将各金属硝酸盐在容量瓶中配制成0.3 mol/L溶液,按照Ce与掺杂离子M为0.92∶0.08的摩尔比量取溶液,将混合溶液充分搅拌,搅拌过程中逐滴加入6 mol/L的NaOH溶液,混合溶液全部变成悬浊液后,调节pH值为12~13。取适量的悬浊液放入反应釜中,将反应釜密封后置于200 ℃的鼓风干燥箱中干燥24 h。反应完成后将反应釜取出、冷却,利用蒸馏水反复抽滤洗涤,于80 ℃下干燥6 h,将干燥好的试样用玛瑙研钵研磨后装袋。
1.2 样品的表征
采用X射线衍射技术(D/Max-2400)在CuKα辐射下测定样品的晶相结构。通过扫描电镜SEM(QUANTA 400)观察样品的形貌。利用装有氦氖离子激光器(532 nm)的拉曼光谱仪(JY-HR800)对样品进行拉曼光谱表征;样品的紫外吸收光谱采用紫外-可见分光光度计(U-3900)测量,扫描范围为200~800 nm,扫描速率为300 nm/min;荧光光谱通过荧光分光光度计(F-4600)测定;样品的氧化还原性能(TPR)的测试是通过PC-1200仪器完成,采用的气体为H2(10%,体积分数,下同)-N2(90%)混合气体,测试温度范围为室温至900 ℃,升温速率为10 ℃/min。
2 结果与讨论
2.1 微观结构表征
图1(a)为纳米Ce0.92M0.08O2(M=Co,Mn,Fe,La)固溶体X射线衍射(XRD)图谱。由图可知,所有样品的衍射峰较为尖锐,表明样品的结晶程度良好,主相均为萤石立方结构的CeO2相(PDF No.65-5923)。其中Co离子掺杂样品含有微量的Co3O4杂相(PDF No.74-2120),表明在该制备条件下,Co离子的固溶限略低于8%。其他掺杂样品均为CeO2相,说明Mn,Fe及La离子全部进入CeO2的晶格中。掺杂离子进入CeO2晶格替代Ce4+后,因掺杂离子半径及其价态与原来的Ce4+不同,会引起晶格发生畸变,进而使XRD峰位角度发生移动。图1(b)为(111) 衍射峰的放大图。与纯CeO2相比,掺杂样品的主峰均发生变化。其中掺杂La及Fe离子样品的峰明显变宽,说明样品的晶粒尺寸变小。可观察到掺杂Mn及Co样品的峰位向高角度移动,特别是Co离子的掺杂移动更为明显。这主要是由于Mn及Co离子半径较小,在相同的掺杂浓度下,离子半径越小,则晶格畸变程度越大,因此峰位移动得更加明显[11-12]。
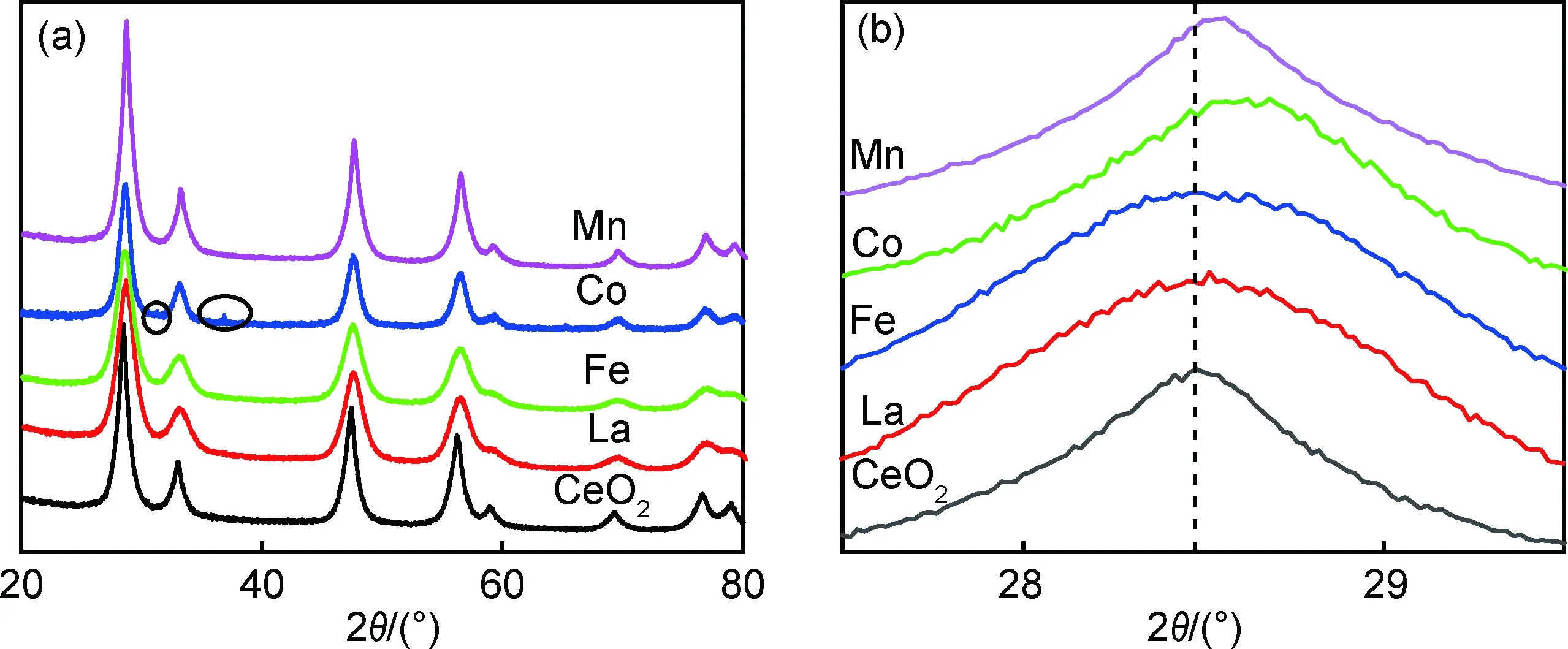
图1 Ce0.92M0.08O2(M=Co,Mn,Fe,La)固溶体X射线衍射图谱(a)和(111)衍射峰放大图谱(b)Fig.1 X-ray diffraction patterns (a) and enlarged patterns of (111) diffraction peaks(b) of Ce0.92M0.08O2(M=Co,Mn,Fe,La) solid solutions
图2(a)为对XRD图谱拟合所得各样品晶胞参数。由图可知,掺杂La离子样品的晶胞参数比纯CeO2略有增大;其他掺杂样品的晶胞参数均低于纯CeO2。掺杂离子进入晶格后,会替换点阵结构中Ce4+的位置,因此不同的离子半径会引起晶格畸变。掺杂离子半径与原Ce4+的半径差异越大,则晶胞参数的变化越为明显。La3+(8配位)离子半径为0.116 nm,大于Ce4+(0.097 nm)及Ce3+(0.1143 nm)的半径,因此La3+的掺杂引起晶胞参数的增大[13];Fe离子的常见价态为Fe2+及Fe3+两种,两种价态的离子半径(rFe2+=0.092 nm,rFe3+=0.078 nm)均小于Ce离子。Co离子通常也具有+2及+3两种价态(rCo2+=0.090 nm,rCo3+=0.061 nm);而Mn离子的价态更加多样,一般具有+2,+3及+4等多种价态(rMn2+=0.096 nm,rMn3+=0.0645 nm,rMn4+=0.053 nm),因此,Fe,Co及Mn离子掺杂均会使CeO2的晶胞参数减小,引起晶格收缩。通过对比可知,半径越小的掺杂离子引起CeO2晶格畸变程度越大,Co离子掺杂引起的晶胞参数收缩程度最为明显,因此推测其掺杂价态应为Co3+,其在CeO2中的固溶限较低。与Co3+掺杂相比,Mn离子掺杂所引起的晶格畸变程度较小,因此离子半径应稍大于Co3+,推测Mn离子的价态应为+2或+3价态。通过谢乐(Scherrer)公式计算样品平均晶粒尺寸d(式(1))。
d=Kλ/(βcosθ)
(1)
式中:K为谢乐常数,为0.89;λ为X射线波长;β为衍射峰半峰宽;θ为衍射角。计算结果如图2(b)所示。样品的晶粒尺寸分别为:纯CeO2(8.9 nm)>Mn(8.5 nm)>Co(8.1 nm)>Fe(5.8 nm)>La(4.9 nm),该结果表明,掺杂离子均可阻止晶粒长大,达到细化晶粒的效果。
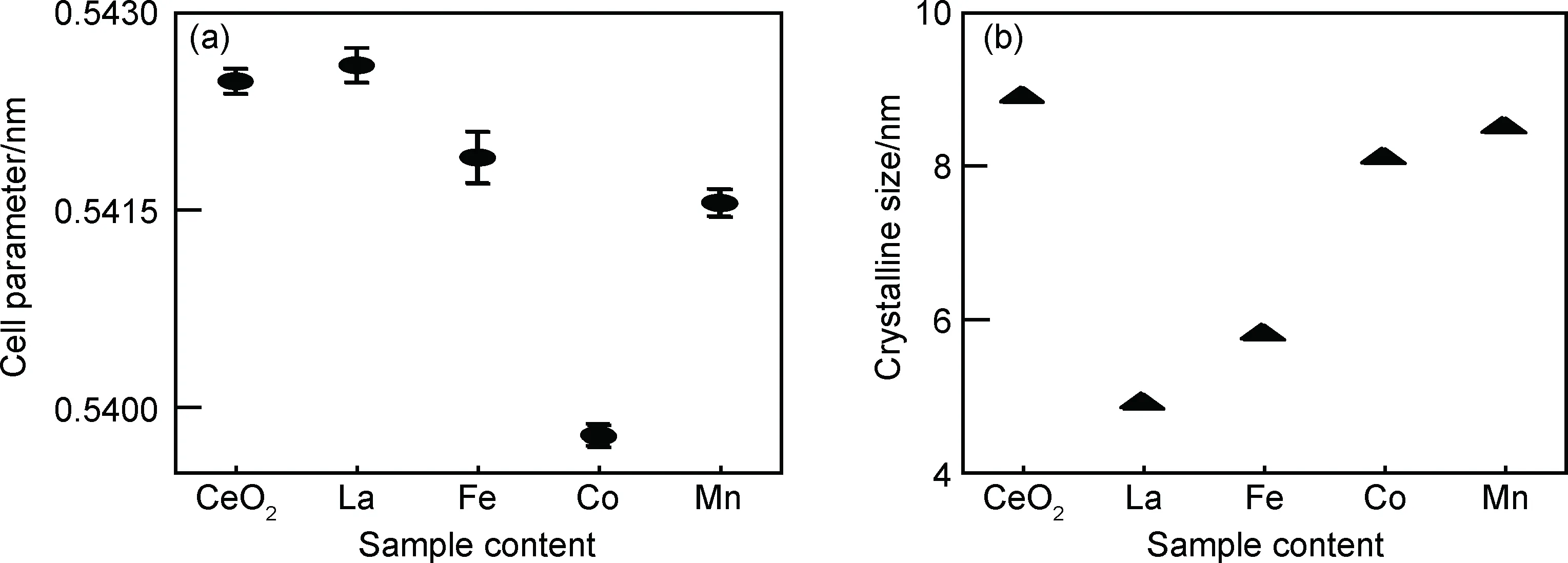
图2 Ce0.92M0.08O2(M=Co,Mn,Fe,La)固溶体晶胞参数(a)和晶粒尺寸(b)Fig.2 Cell parameters(a) and crystalline size(b) of Ce0.92M0.08O2 (M=Co,Mn,Fe,La) solid solutions
2.2 微观形貌分析
通过扫描电镜进一步了解样品的晶粒尺寸及形貌特征。图3为典型样品Ce0.92Mn0.08O2及Ce0.92Fe0.08O2样品的扫描电镜图。可知样品均显示为球形,且大小分布均匀,但由于样品的晶粒尺寸较小,表面能高,因此团聚较为严重。样品的晶粒尺寸约为10 nm,该结果与XRD计算结果相一致。

图3 典型样品扫描电镜图 (a)Ce0.92Mn0.08O2;(b)Ce0.92Fe0.08O2Fig.3 SEM images of typical samples (a)Ce0.92Mn0.08O2;(b)Ce0.92Fe0.08O2
2.3 紫外光谱分析
离子掺杂会改变CeO2的电子跃迁情况,图4(a)为Ce0.92M0.08O2(M=Co,Mn,Fe,La)固溶体的紫外吸收光谱。如图所示,所有样品在200~400 nm之间都存在一个较强且宽的吸收峰,其中240 nm对应于从O2+到Ce4+之间的电荷转移,290 nm和340 nm处的峰代表着从O2+到Ce4+之间的电荷转移和带间跃迁[14]。掺杂使样品的吸收边都发生了不同程度的红移[15]。特别是过渡金属离子掺杂的样品,红移程度尤为明显。对于Mn及Co离子掺杂样品,在400~800 nm范围内存在吸收,表明该区域存在掺杂离子的跃迁[16]。图4(b)为通过固溶体的紫外吸收光谱按照直接跃迁模式拟合所得到的能隙图。由图可知,各样品能隙值从大到小的排列为CeO2>La>Fe>Co>Mn。影响能隙值的原因包括样品的晶粒尺寸、晶格内杂质离子形成的新能级以及氧空位形成的能级等。最主要的原因在于掺杂离子可在CeO2的导带下形成离散的空能级,允许电子从CeO2的价带跃迁到较低的杂质能级上[17],从而使得能隙红移[18]。La和Ce同为镧系元素,离子半径及化学性质相似,因掺杂所引起的晶格畸变小,因此能隙值比纯CeO2只是稍有降低。其他三种过渡金属离子的半径小、价态低,因此引起晶格畸变大,且因离子价态低,使得晶格内部因电荷补偿而产生更多氧空位,从而使固溶体的能隙降低较为明显。对比Fe,Co及Mn离子的掺杂样品可知,Mn离子对材料能隙值的影响最为显著,这与其呈现多种价态且离子半径较小有关。
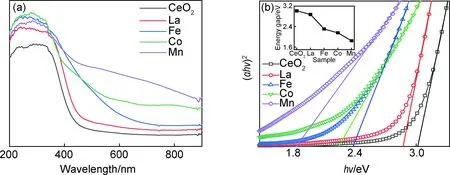
图4 Ce0.92M0.08O2(M=La,Fe,Co,Mn)固溶体的紫外吸收光谱(a)和紫外能隙拟合图(b)(插图为各样品的能隙值)Fig.4 UV absorption spectra(a) and fitting patterns of band gap(b) (the inset shows the band gap energy values) of Ce0.92M0.08O2 (M=La,Fe,Co,Mn) solid solutions
2.4 荧光光谱分析
通常,荧光光谱发射峰(PL)的强度能够反映受激电子和空穴的复合率,即较高的发射峰强度表明电子-空穴对的复合率较高。图5为Ce0.92M0.08O2(M=Co,Mn,Fe,La)固溶体在激发波长为325 nm条件下的荧光光谱。由图5可知,所有样品的发射峰形状相似。最强峰位于468 nm,还有分别位于422,439,451,484,506,530 nm和581 nm等多个强度较弱的峰。这几个峰之间互相重叠,形成了从370 nm到600 nm范围内的宽峰。这些发射峰是由于在Ce4f与O2p之间存在多个晶格缺陷能级,不同能级之间跃迁所形成的[18]。其中430 nm到540 nm范围内的发射峰对应于由不同缺陷能级跃迁至价带的跃迁,从540 nm至560 nm间的发射峰则源于晶格中的氧空位[19-20]。通过对比强度可知,各样品的峰强度差别很大。纯CeO2具有最高强度的发射峰,当Co,Mn,Fe和La离子掺入氧化铈晶格后,发射峰的强度急剧下降,掺杂使材料中氧空位和晶格缺陷的浓度增加,因此会延迟受激电子和空穴的复合过程,从而导致光致发光峰的强度降低[21]。不同掺杂离子对样品的发射峰强度影响不同,各样品峰强度由大到小依次为:CeO2>La>Fe>Co>Mn。该结果表明,不同掺杂离子对CeO2晶格内部影响不同,荧光峰的强度越低,说明晶格内的氧空位及缺陷浓度越高,该结果与紫外分析结果相一致。
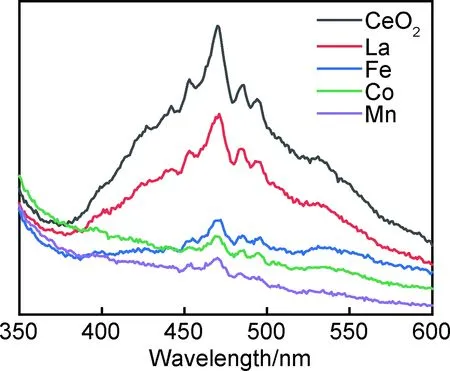
图5 Ce0.92M0.08O2(M =Co,Mn,Fe,La)固溶体的荧光光谱Fig.5 PL spectra of Ce0.92M0.08O2 (M=Co,Mn,Fe,La) solid solutions
2.5 Raman图谱分析
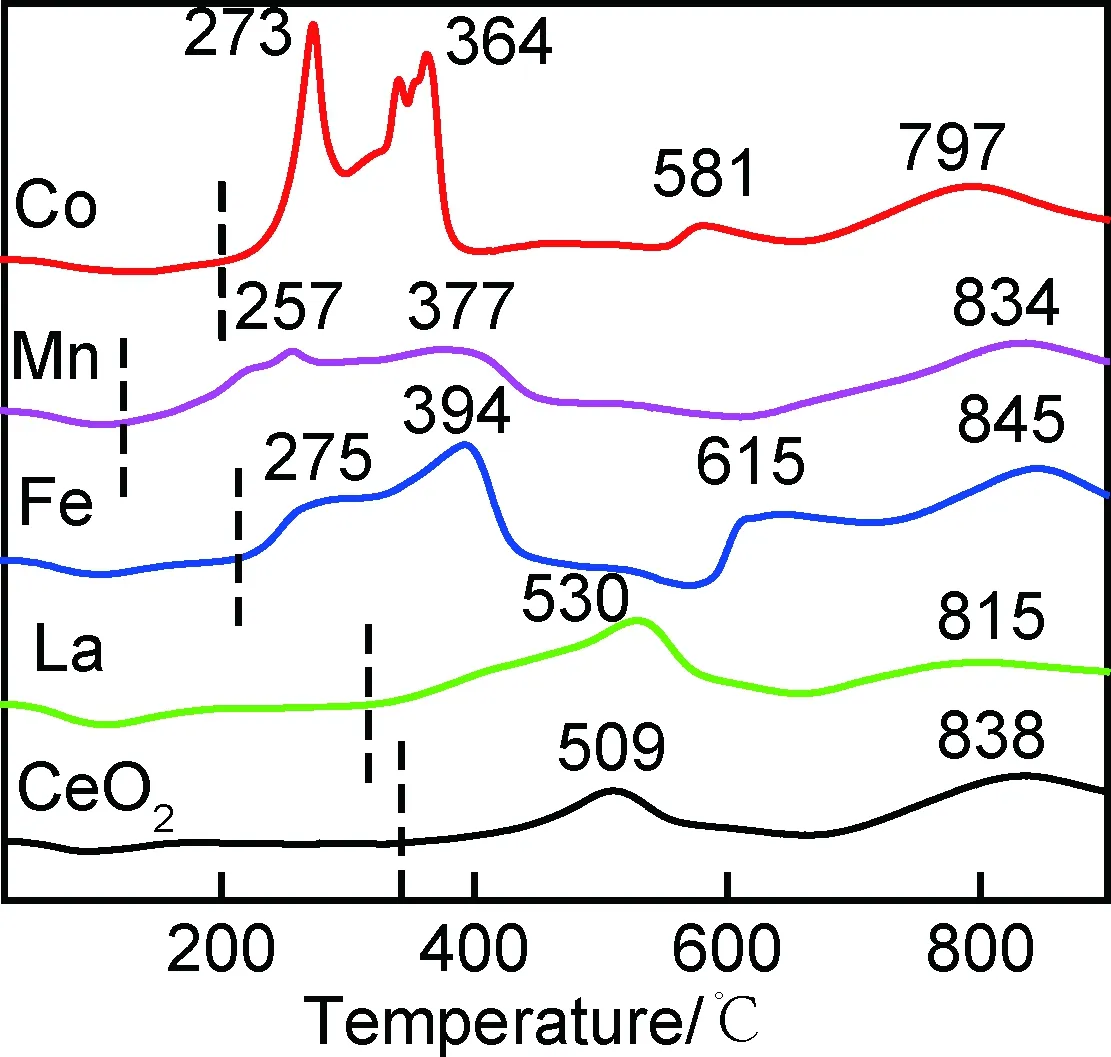
CeO2晶格中掺杂离子的存在会引起晶格结构发生畸变,通过Raman光谱可快速准确地探测样品微观结构变化情况。CeO2为萤石立方结构,这种结构的拉曼光谱受氧晶格振动支配[22]。图6(a)为Ce0.92M0.08O2(M=Co,Mn,Fe,La)固溶体的Raman图谱。由图可知,在460 cm-1的强峰是对应于萤石立方结构的F2g对称振动,该模式源于金属离子与氧(M—O)的对称振动模式[23]。与纯CeO2相比,掺杂样品的F2g特征峰强度均发生了降低,且峰形变宽,对称性降低,这主要是由于样品的晶粒尺寸以及掺杂引起的晶格缺陷增大[24]。其中掺杂Co样品在530 cm-1处存在一个对应于Co氧化物杂相的拉曼峰,而其他固溶体的拉曼图谱均与纯CeO2峰形相同,表明除Co离子外,其余掺杂离子均溶入CeO2晶格中形成了固溶体。图谱中位于600 cm-1附近的峰对应的是氧空位特征峰[25],该峰强度越高,则代表氧化物晶格中氧空位浓度越高。本工作通过氧空位特征峰D与F2g特征峰面积比值ID/IF2g来衡量固溶体中氧空位相对浓度的大小,结果如图6(b)所示,氧空位浓度由小到大的顺序依次为:CeO2 图6 Ce0.92M0.08O2(M=Co,Mn,Fe,La)固溶体的拉曼图谱(a)和ID/IF2g比值图(b)Fig.6 Raman spectra(a) and ID/IF2g ratio graph(b) of Ce0.92M0.08O2(M=Co,Mn,Fe,La)solid solutions 经过掺杂修饰可在CeO2晶格中引入更多的氧空位,从而有效增强氧离子的扩散性,提高CeO2的还原能力。不同掺杂离子对CeO2还原性的影响能力通过TPR测试进行分析,图7为各样品在室温至900 ℃范围内与H2的程序升温还原(TPR)图。图中峰值对应于样品与H2发生最大还原反应速率所对应的温度,而还原峰的积分面积则对应于样品与H2的反应量。纯CeO2有两个还原峰,第一个峰起始于340 ℃(如虚线所示),峰值位于509 ℃,对应于CeO2表面的Ce4+还原为Ce3+,另一个还原峰值出现在838 ℃,属于体相内Ce4+的还原峰[26-27]。表面还原过程中,氧原子从CeO2的表面移除并形成了氧空位。随着还原反应的进行,这些空位将逐渐从表面迁移至体相内,而氧原子则从体相向表面扩散。La掺杂样品的表面还原峰起始于315 ℃,峰值位于530 ℃。尽管峰值温度高于纯CeO2,但还原反应的起始温度有了降低,且表面还原峰积分面积增大,对应于与氢的还原量提高,因此掺La固溶体的还原能力得到了一定提高。掺杂Fe,Mn及Co样品的还原温度明显降低,其起始还原温度分别位于213,123 ℃和200 ℃;表面还原峰是由两个或多个部分重叠的还原峰组成,特别是Co掺杂固溶体因有Co氧化物杂相的存在,因此还原峰更为复杂。此外,掺杂Fe和Co样品还在615 ℃和581 ℃处也存在还原峰。CeO2中掺杂过渡金属离子后,与H2发生还原反应变得较为复杂,除了存在Ce4+的还原外,同时还有多价态过渡金属离子的还原,因此掺杂Fe,Mn及Co样品的还原峰数目变多,还原起始温度及峰值温度均明显降低,表明固溶体还原活性得到了显著提升[28]。通过比较各样品的体相还原峰值温度可知,掺杂离子对固溶体体相还原温度的影响各不相同。掺杂La样品的体相还原峰值温度降低,掺杂Fe样品的体相还原峰值温度略微增大,而Mn及Co掺杂样品的还原峰值温度稍有降低。该结果表明掺杂过渡金属离子主要影响固溶体的表面还原特性,而对体相还原影响不明显。进一步比较各样品还原峰的积分面积,得出各样品消耗H2量由小到大依次为CeO2 图7 Ce0.92M0.08O2(M=Co,Mn,Fe,La)固溶体的H2-TPR图Fig.7 H2-TPR patterns of Ce0.92M0.08O2(M=Co,Mn,Fe,La) solid solutions (1)除掺杂Co样品出现杂相外,其余掺杂固溶体均为CeO2纯相,固溶体的晶胞参数变化趋势与掺杂离子半径相一致。 (2)掺杂离子引起CeO2晶格畸变,氧空位浓度增大,氧空位浓度由小到大的顺序依次为CeO2 (3)掺杂样品与H2反应的还原温度降低,与氢的反应能力提高。掺杂离子价态及离子半径与Ce离子差异越大,则越有利于提高固溶体与H2的还原反应活性。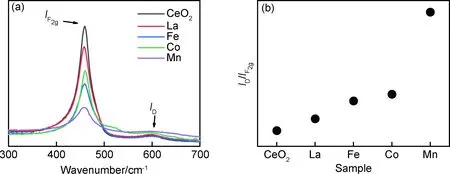
2.6 氧化还原性能分析
3 结论
