地西他滨联合芦可替尼治疗老年不典型慢性粒细胞白血病:1例报道并文献复习
2023-03-18娄典刘利秦炜炜
娄典,刘利,秦炜炜
空军军医大学第二附属医院血液内科,陕西 西安 710038
不典型慢性粒细胞白血病(atypical chronic myeloid leukemia,aCML)是一种很罕见的BCR-ABL融合基因阴性的骨髓增生异常综合征/骨髓增殖性肿瘤(myelodysplastic syndrome/myeloproliferative neoplasm,MDS/MPN)亚型,其特征为中性粒细胞增多伴前体细胞增加、粒系发育异常,以及外周血和骨髓中的原始细胞<20%[1]。基于地西他滨在治疗MDS和慢性粒单核细胞白血病(chronic myelomonocytic leukemia,CMML)中取得了良好疗效,有研究者已尝试将其用于aCML的治疗。2013年Maxson等[2]首次在aCML和慢性中性粒细胞白血病(chronic neutrophilic leukemia,CNL)中发现CSF3R突变,且体外实验结果显示CSF3R膜近端突变T618I可能对JAK抑制剂芦可替尼敏感。目前国内外尚无地西他滨联合芦可替尼治疗aCML的报道。本文报告1例地西他滨联合芦可替尼方案治疗老年aCML的临床疗效,并结合病例资料进行相关文献复习,旨在帮助临床医师提高对该病的认识。
1 病例资料
1.1 病史 患者,男,72岁,因“乏力、头晕1个月余”入院。2020年4月开始无明显诱因出现乏力、头晕,活动后加重,伴明显盗汗,无发热、咳嗽等不适,就诊于当地医院,查血常规:白细胞66.73×109/L,血红蛋白132 g/L,血小板67×109/L,加用“羟基脲 1.5 g 3次/d”口服降白细胞治疗后,患者自觉症状较前无好转,2020年5月11日为明确血细胞异常的病因就诊于空军军医大学第二附属医院,并收治入血液内科。
1.2 入院后查体 体温36.8 ℃,脉搏90次/min,呼吸20次/min,血压125/75 mmHg,体型偏胖,自主体位。全身皮肤无黄染及淤斑,浅表淋巴结未触及肿大,胸骨无压痛,心肺查体未见异常,腹软、无压痛,肝脾肋缘下未扪及,双下肢轻度水肿。
1.3 辅助检查 血常规:白细胞74.17×109/L,中性粒细胞比例61.9%,单核细胞比例7.5%,淋巴细胞比例14.4%,嗜酸性粒细胞比例15.8%,嗜碱性粒细胞比例0.4%,血红蛋白108 g/L,血小板40×109/L。乳酸脱氢酶342 U/L;红细胞沉降率16 mm/h;肝肾功能、电解质、降钙素原、C反应蛋白、免疫球蛋白补体系列及自身抗体ANA谱均正常。
1.4 影像学及其他检查 腹部超声示:肝脏大小正常,脾脏厚度5.1 cm;胆、胰、双肾未见明显异常。骨髓细胞形态学检查显示:增生极度活跃,粒系占96.0%,红系占2.5%;粒系增生极度活跃,其中原始粒细胞占14.0%,可见双核中幼粒细胞、双核晚幼粒细胞、环形核,个别晚幼核粒细胞胞质颗粒减少,嗜酸性粒细胞占16.5%,未见嗜碱性粒细胞;红系增生减低;淋巴细胞形态未见异常;全片见巨核细胞45个;中性粒细胞碱性磷酸酶(neutrophils alkaline phosphatase,NAP)阳性率为83%,积分值173分。外周血涂片分类示:白细胞增高,原始粒细胞占10.0%,嗜酸性粒细胞占13.0%。骨髓活检显示增生活跃,粒系增生明显活跃,原始粒细胞比例增高,嗜酸性粒细胞比例增高;红系增生减低,巨核细胞少。骨髓病理检查显示:CD34(+)散在,CD68(+)偶见,CD61(–),符合髓系肿瘤,原始及幼稚细胞比例占10%(图1)。骨髓细胞流式免疫分型示:原始/幼稚髓细胞占有核细胞总数的11.8%,表型为CD34、CD117、CD33,部分表达HLA-DR、CD13、CD7、CD56、CD19、CD10、CD3、CD5、CD2、CD41,粒细胞相对比例增高,其免疫表型CD11b、CD16、CD13、CD15未见明显表达紊乱。包含BCR-ABL(P190、P210和P230)、FIP1L1-PDGFRA、TEL-PDGFRB、KIF5B-PDGFRA、BCR-PDGFRA、ETV6-PDGFRA、STRN-PDGFRA、CDK5RAP2-PDGFRA和TEL-JAK2等在内的融合基因检测结果均为阴性。染色体核型:46,XY[20]。荧光原位杂交(fluorescence in situ hybridization,FISH)未检测出+8、–5/5q–、–7/7 q–、2 0 q–、–Y 等异常核型及P D G F R A、PDGFRB、FGFR1和JAK2基因重排。应用二代测序技术对34种常见髓系肿瘤基因突变进行筛查,结果显示检测到CSF3R基因突变,突变位点为Exon14,核苷酸改变c.1853C>T,氨基酸改变p.T618I,突变频率47.15%,未检测到JAK2、MPL、CALR、SETBP1、ASXL1、N/K-RAS、SRSF2和TET2等其他基因突变。

图1 伴CSF3R突变aCML患者的骨髓病理结果(HE)Fig.1 Pathological examination of bone marrow in aCML patient with CSF3R mutant (HE)
1.5 诊断、治疗及随访 患者为老年男性,外周血白细胞明显增多,以中性粒细胞及其前体细胞(早幼粒细胞、中幼粒细胞、晚幼粒细胞)增多为主,其中前体细胞占白细胞的比例>10%,合并双核中幼粒细胞、双核晚幼粒细胞及环形核等明显的粒系发育不良特征,外周血和骨髓中原始细胞<20%,无Ph和BCR-ABL融合基因,结合患者病史、体征诊断为aCML。入院后治疗方案:该患者无异基因造血干细胞移植(allogeneic hematopoietic stem cell transplantation,allo-HSCT)指征,给予地西他滨治疗:20 mg/(m2.d),第1~5天,4~8周/次,共4个疗程。治疗过程中患者血常规指标变化如图2所示。第1个疗程骨髓造血恢复后复查骨髓细胞形态学示达到完全缓解(complete remission,CR),嗜酸性粒细胞比例不高;复查骨髓病理示原始细胞比例不高(图3)。地西他滨治疗2个疗程后复查流式微小残留病变(minimal residual disease,MRD)转阴。该患者存在CSF3R T618I突变,故在第1个疗程中细胞恢复后给予芦可替尼30 mg/d靶向治疗,并依据患者血小板计数调整剂量。芦可替尼治疗4个月后采用二代测序监测分子谱显示,CSF3R T618I突变负荷降至2.02%。患者全部治疗结束后继续口服芦可替尼单药维持治疗。安全性方面,患者仅在第1个疗程的地西他滨治疗后合并Ⅳ度骨髓抑制,予以刺激骨髓造血、输注血制品、预防感染等支持治疗后好转,第2~4个疗程地西他滨联合芦可替尼治疗过程中仅合并Ⅰ-Ⅱ度骨髓抑制,整个治疗过程中无非血液学不良事件发生。随访至2021年12月,患者无不适,监测血常规正常,骨髓细胞形态持续为CR状态,MRD持续阴性,CSF3R T618I突变负荷<5.00%。
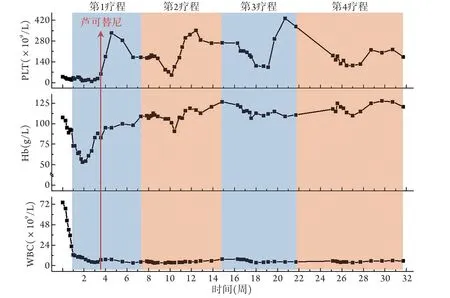
图2 aCML患者治疗过程中白细胞、血红蛋白和血小板变化情况Fig.2 Changes of leukocytes, hemoglobin and platelets of the case during treatment process

图3 aCML患者地西他滨治疗1个疗程后骨髓病理检查结果(苏木精-吉姆萨-酸性品红染色)Fig.3 Pathological examination of bone marrow of aCML patient after one course of treatment with decitabine (Hematoxylin-Giemsa-Acid fuchsin staining)
2 文献检索及复习
以中文“不典型慢性粒细胞白血病”“地西他滨”“阿扎胞苷”“芦可替尼”“去甲基化药物/去甲基化治疗”为关键词,检索中国知网和万方数据知识服务平台数据库自建库至2021年12月报道的有关aCML的文献,筛查到1篇有关地西他滨治疗aCML的中文个案报道[3];以英文“atypical chronic myeloid leukemia”“decitabine”“azacitidine”“ruxolitinib”“hypomethylating agents”为关键词,检索PubMed数据库自建库至2021年12月报道的有关aCML的文献,共检索到38篇英文文献,经获取全文后,共筛选出6篇去甲基化药物治疗aCML的文献[4-9],4篇有关芦可替尼治疗aCML的文献[10-13],均为个案报道。
文献报道应用去甲基化药物地西他滨或阿扎胞苷治疗aCML的患者共11例[3-9](表1),其中采用单药地西他滨治疗aCML的患者共8例[3-6],地西他滨的给药剂量为20~25 mg/m2,1次/d,共5 d,大部分患者的治疗周期为4~6周,仅1例患者的治疗周期为1~3个月,中位疗程数为4(1~6)个,大部分患者可获得良好的血液学反应,CR率可达87.5%(7/8)。采用单药阿扎胞苷治疗aCML的患者共2例[7-8],1例经过4个疗程的单药阿扎胞苷诱导治疗后脱离输血依赖,另1例患者先后使用(聚乙二醇)干扰素、达沙替尼、羟基脲、芦可替尼和白消安治疗后不耐受或治疗失败,患者仍依赖反复输血,伴脾脏肿大和白细胞增高,4年半后转为急性髓系白血病(acute myeloid leukemia,AML),二代测序检测到FLT3内部串联重复突变,采用DA方案诱导治疗失败后换用阿扎胞苷联合索拉非尼治疗,疗效评估为部分缓解(partial remission,PR),且患者仍未脱离输血依赖。文献还报道1例采用地西他滨联合CAG方案化疗的aCML患者,疗效评价为CR[9]。文献[10]-[13]报道的采用芦可替尼单药治疗的aCML患者共4例,均伴有CSF3R T618I突变,2例治疗有效,2例无效(表2)。
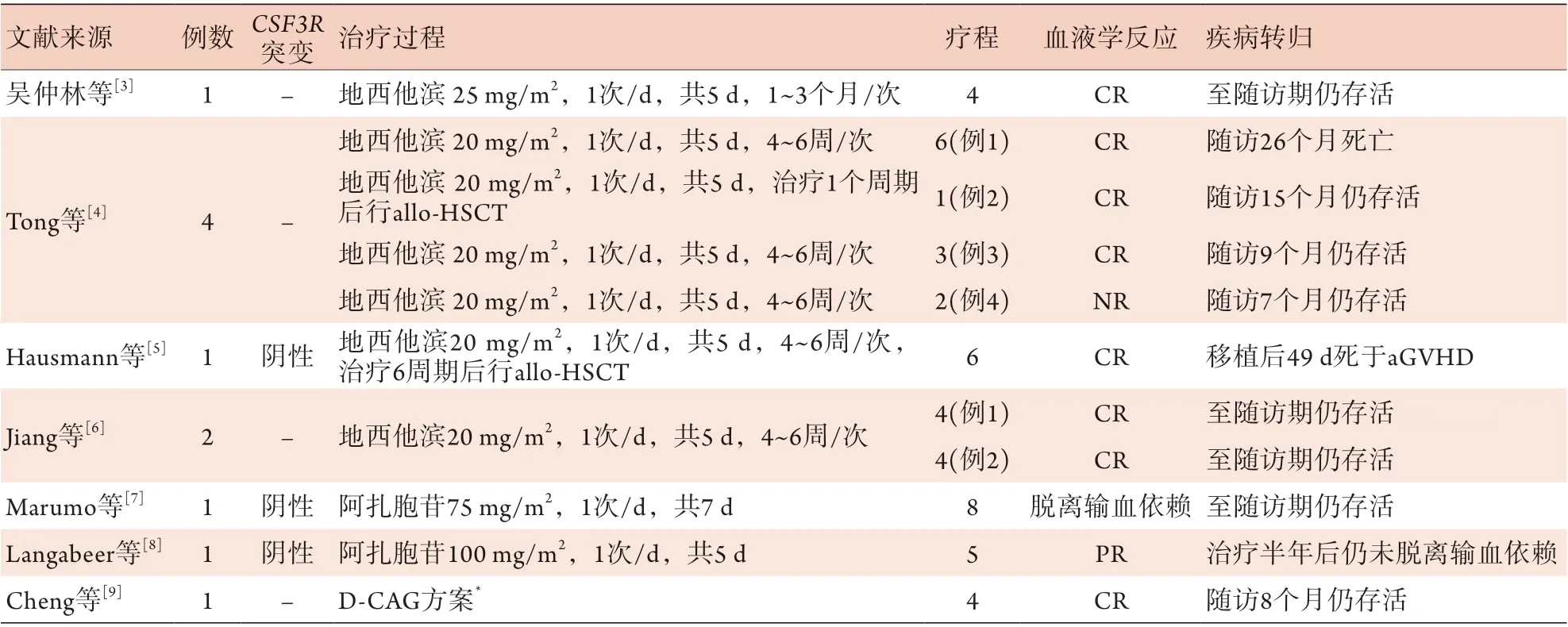
表1 文献报道的去甲基化药物治疗aCML的情况Tab.1 The aCML treated with demethylated drugs reported in previous literature
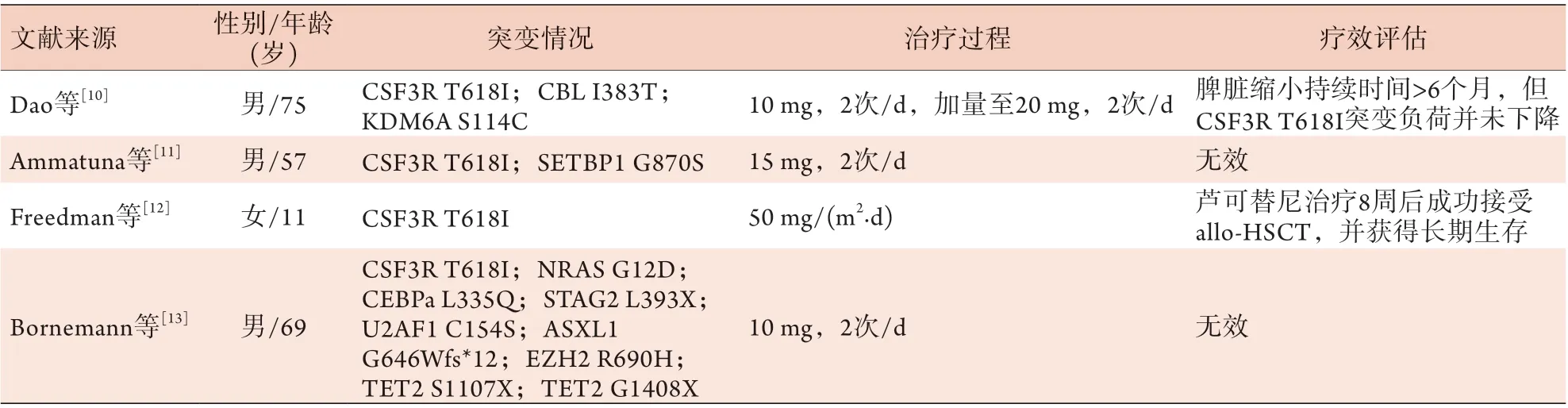
表2 文献报道的芦可替尼治疗伴CSF3R T618I突变aCML的情况Tab.2 The aCML with CSF3R T618I mutant treated with ruxolitinib reported in previous literature
3 讨 论
aCML的发病年龄大多在60岁左右,约半数患者可合并肝脾大、白细胞计数增高、贫血和血小板减少[14]。由于发病率低,迄今为止尚未检测到与aCML相关的特异性分子异常,该病的临床诊断仍较困难。因此,最新的WHO 2016版aCML的诊断标准[1]仍基于形态学基础:(1)外周血白细胞增多,由中性粒细胞及其前体细胞(早幼粒细胞、中幼粒细胞、晚幼粒细胞)增多导致,前体细胞在白细胞计数中≥10%;(2)无Ph和BCR-ABL融合基因;(3)粒细胞生成异常(包括染色质凝集异常);(4)无或轻微嗜碱性粒细胞增多(<2%);(5)无或轻微单核细胞绝对值增多(<10%);(6)骨髓粒系增生明显活跃,粒系发育异常,伴或不伴红系或巨核系发育异常;(7)外周血和骨髓中原始细胞<20%;(8)无PDGFRA、PDGFRB、FGFR1重排,无PCM1-JAK2;(9)不符合WHO规定的慢性粒细胞白血病(chronic myeloid leukemia,CML)、原发性骨髓纤维化(primary myelofibrosis,PMF)、真性红细胞增多症(polycythemia vera,PV)或原发性血小板增多症(primary thrombocytosis,ET)的诊断标准。本例患者二代测序检测到CSF3R突变,目前研究者普遍认为该突变在CNL中更常见[2,15],也有研究发现,其在aCML中的突变率约为26%,但其导致aCML的具体机制尚不明确[16]。aCML与CNL有很多重叠的临床特征,参照WHO的诊断标准,aCML与CNL最主要的区别是CNL外周血前体细胞占白细胞的比例<10%,无粒系发育异常,且原始细胞<5%,因此,本例患者虽然检测到CSF3R突变,但不符合CNL的诊断标准。此外,本例患者起病时外周血和骨髓中嗜酸性粒细胞比例明显增高,但未检测到PDGFRA、PDGFRB、FGFR1或PCM1-JAK2相关的分子学及遗传学异常,因此,不符合伴嗜酸性粒细胞增多的髓系肿瘤的诊断标准[1]。
aCML是一种罕见疾病,目前国内外尚缺乏标准的治疗方案。当患者出现进行性白细胞增多、贫血或血小板减少、脾大或出现与疾病相关的症状时应及时治疗,对于有合适供者的年轻患者建议首选allo-HSCT,但aCML患者移植的最佳时机仍然存在争议[17]。依据文献报道,对于有治疗指征但不适合移植的患者,可考虑参加临床试验,但由于其发病率低,目前大型随机临床试验数量少,且入组患者多为急变期或白血病期患者,故临床试验常不可及[17-18]。鉴于aCML与其他慢性髓系肿瘤具有许多共同特征,其治疗策略可参照MDS或MPN,加用羟基脲或干扰素等药物来降低肿瘤负荷,并缓解脾大的症状,但对进展期患者疗效不佳甚至无效。基于去甲基化药物地西他滨或阿扎胞苷在CMML中的应用取得了较好的疗效[19],已有研究尝试将这些药物用于aCML的治疗[3-9]。迄今为止,文献报道应用单药地西他滨治疗aCML的CR率可达87.5%,但长期生存能否获益仍需通过更大样本量及更长时间的随访观察来证实[3-6]。本例患者高龄,血常规提示白细胞明显增多,合并贫血和血小板减少,有治疗指征,但不具备移植或临床试验的条件,而加用羟基脲降白细胞的效果有限,为延缓疾病进展及改善患者的生存状况,给予地西他滨方案诱导及巩固治疗,1个周期后即获得了良好的血液学反应,达到形态学CR状态,2个周期后MRD转阴,动态监测MRD持续阴性,且患者耐受性良好。
随着分子诊断技术的不断发展,越来越多的突变基因在aCML中被检测到,对aCML的认识也越来越深入,对其治疗也具有指导意义。aCML患者较常合并SEBTP1、ASXL1、N/K-RAS、SRSF2和TET2基因突变,也可合并CBL、CSF3R、JAK2、EZH2和ETNK1基因突变,但发生率较低[20]。其中一些突变基因可影响酪氨酸蛋白激酶-信号转导及转录激活因子(Janus kinase-signal transducers and activators of transcription,JAK-STAT)、丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)或Rho相关的卷曲蛋白激酶(Rho-associated coiled-coil protein kinase,ROCK)信号通路[21-23],如NRAS、JAK2和CSF3R突变可直接影响MAPK和JAK-STAT通路,SETBP1编码一种名为SET结合蛋白1(SEB)的蛋白质,后者可影响MAPK通路;针对相应突变位点的靶向药物可抑制这些异常的信号通路,为不适合移植的患者提供了新的治疗手段。CSF3R基因是粒细胞集落刺激因子的受体,在粒细胞的生长和分化中起重要作用。CSF3R突变主要分为两类:一类是导致受体胞质尾部截断的无义或移码突变,另一类是膜近端的点突变,它们分别导致下游的原癌基因酪氨酸蛋白激酶Src(proto-oncogene tyrosineprotein kinase Src,SRC)或JAK激酶活化而使细胞增殖。体外研究发现,截断突变对SRC激酶抑制剂达沙替尼敏感,而膜近端突变可激活JAK-STAT通路,对JAK激酶抑制剂芦可替尼敏感[2]。膜近端的点突变T618I为其热点突变位点,在小鼠模型中JAK激酶抑制剂芦可替尼能有效抑制CSF3R T618I驱动的恶性细胞增殖,并能减轻突变引起的白细胞增高和脾大症状[24]。但临床上应用芦可替尼治疗aCML的经验仍不足,有报道其对伴CSF3R T618I突变的aCML的有效率为50%,患者应用芦可替尼治疗后若能桥接移植可获得长期生存[10-13]。一项针对芦可替尼治疗CNL和aCML的安全性和有效性的Ⅱ期临床研究(NCT02092324)结果显示,患者的总体有效率为32%,其中伴有CSF3R突变组的有效率为54%,进一步扩大样本量的研究结果显示,与PR组及无应答组比较,CR组患者在6个周期治疗后的CSF3R T618I平均突变负荷明显降低,除34%和14%的患者分别出现贫血和血小板减少的3级血液学毒性外,未观察到与芦可替尼相关的严重不良事件[16]。本例患者检测到CSF3R T618I突变,在使用芦可替尼治疗4个周期后CSF3R T618I突变负荷下降>90%,提示获得了分子学缓解,为患者长期生存提供了可能;且地西他滨联合芦可替尼方案未增加治疗相关的血液学毒性,无非血液学不良事件发生,提示患者对该联合方案耐受性良好。
到目前为止,allo-HSCT仍是唯一可能治愈aCML的手段,分子靶向治疗也为患者桥接移植奠定了基础,然而由于aCML罕见且多为老年患者,因此适合接受移植治疗的患者极少。Koldehoff等[25]报道21例aCML接受allo-HSCT后的5年总生存率为80%,中位生存期为48个月。Onida等[26]报道42例aCML患者接受allo-HSCT后随访89个月,87%的患者仍处于CR状态,5年总生存率为51%,无复发生存率为36%。但目前对于移植的最佳时机仍存在争议,Gotlib[17]建议将allo-HSCT作为aCML的一线治疗方案,而Talati等[18]则建议根据患者年龄、白细胞计数及分子学异常等危险因素进行危险分层后再决定是否行移植治疗。总之,对于移植指征的把握,临床医师应根据患者的年龄、体能状况、是否有合适供者及是否存在靶向突变等因素来综合评估,以选择最佳的治疗方案。
aCML虽然发病率低,但预后极差,已有研究显示其中位总生存期为12~25个月,AML转化率约为40%,中位转化时间为11.2个月,影响患者生存的不良预后因素包括年龄>65岁、女性、白细胞计数>50×109/L、合并SETBP1突变及外周血中不成熟前体细胞增多等,若患者合并明显的肝脾肿大、单核细胞增多、骨髓原始细胞>5%、明显的红系病态及输血依赖等,则进展为AML的风险更高[21,27-29]。本例患者具有高龄、起病时白细胞计数增多、外周血及骨髓中原始细胞比例偏高等不良预后因素,预后较差。因此,笔者采用地西他滨联合靶向药物芦可替尼治疗,患者已获得CR,且MRD早期转阴,CSF3R T618I突变负荷大幅下降,至随访截点,患者的生存期达19个月,预后明显改善。笔者后续将继续评价芦可替尼在维持治疗中的疗效。
综上所述,aCML诊断较困难,临床上仅依靠形态学改变可能造成一定的漏诊,随着分子生物学技术的不断发展,MDS/MPN相关的分子标记物不断被检出,加深了我们对aCML的认识。虽然CSF3R T618I突变不是aCML特异的分子标记物,但针对该突变位点的靶向治疗可明显改善患者的预后。本例患者72岁,无移植指征,采用地西他滨联合芦可替尼方案治疗后获得了显著的临床疗效,血液学反应和分子学反应均良好,且安全性高,但其远期临床结局仍待进一步追踪评价。
