Development and Characterization of Microsatellite Markers for Harpadon nehereus Based on High-Throughput Sequencing and Cross-Species Amplification in Three Myctophiformes Fishes
2023-03-17HUANGXinxinNINGZijunandYANGTianyan
HUANG Xinxin, NING Zijun, and YANG Tianyan
Development and Characterization of Microsatellite Markers forBased on High-Throughput Sequencing and Cross-Species Amplification in Three Myctophiformes Fishes
HUANG Xinxin, NING Zijun, and YANG Tianyan*
,,316022,
is a widespread economical fish found in the coastal seas of China and has important ecological va- lue in the marine ecosystem. However, its germplasm resources have been seriously degraded due to natural factors and anthropo- genic activities. In this study, high-throughput sequencing was applied to search for microsatellite loci intranscriptome to provide references for its resource conservation and utilization. Polymorphic loci were developed by non-denaturing polyacryla- mide gel electrophoresis, and their cross-species amplified ability was detected in three related species. A total of 5652 microsatellites were identified from 16974320 unigenes. Among the primer pairs designed for 100 SSRs for PCR amplification, 80% were success- fully amplified, and 26 loci were polymorphic with a high number of alleles from 3 to 11 each. The expected (e) and observed(o) heterozygosities were 0.355–0.885 and 0.375–0.958, respectively. Most of the loci were highly polymorphic (polymorphism infor- mation content: 0.316–0.852; mean: 0.713), and these markers can be applied in the population genetic diversity research of. However, the transferability of these primers was low, probably because of the close relation of the collected species. In fol- low-up work, simple sequence repeats will be excavated with genome-based technologies, and related species will be gathered to address the present inadequacies.
; microsatellite markers; high-throughput sequencing; cross-species amplification
1 Introduction
belongs toMyctophiformes order, Harpadontidae family, andgenus, and lives off-shore on the mud and sandy bottoms of the Indian and Wes- tern Pacific Oceans (Chen, 2002). Owing to its abundant re-sources, delicious taste, and value-added meat quality, thisfishhas become a commercially and ecologically impor- tant species in South and Southeast Asia countries (Zhang., 2009; Zhu., 2014; Nugroho., 2015). How- ever, overfishing, habitat destruction, and marine pollutionhave resulted in the small size and early maturation ofin various sea areas (Hasin, 2016; He., 2018; Guo., 2019a). The declining production ofhas been reported as early as 2017 (Taqwa., 2020). Thus, the protection and rational utilization offishery resources are urgent.
One of the most efficient ways to protect a species is topreserve its genetic resources and diversity (Hedrick, 2001).Previous researches onmainly focused on bio-chemical composition, feeding habits, population dynamics, and food processing (Zhang and Jin, 2014; Zhu., 2014; Sarker., 2017; Chakraborty., 2020); However, only limited references regarding its population genetics are available. SRAP results showed that the population ofin the southeast coast of China exhibited re- markably geographical divisions, stable regional genetic structure, and low genetic diversity level (Zhu., 2014). Population genetic studies based on mitochondrial Cytand2 genes indicated that the genetic variation ofmainly occurs in individuals within populations (Guo., 2019b; Jiang., 2020). An increasing number of reports inferred the genetic characteristics among popu- lations by conducting a combined analysis of different DNA regions (Lu., 2001). Therefore, different methods for further genetic research on, including popula- tion genetics, are necessary.
In the study of germplasm genetic diversity, DNA mar- kers are the most reliable technique in distinguishing allelevariations (Jang., 2020). Microsatellites, also referredas simple sequence repeats (SSRs), are groups of tandem- ly repeated DNA sequences with a few nucleotides (gene- rally 1–6 repeat units) (Zane., 2002; Chistiakov.,2006). As the representative of second-generation molecu- lar markers, SSRs are suitable for the mapping of econo- mically important quantitative traits, breeding improvement, linkage map construction, and population genetic research of aquatic organisms due to their advantages of rich poly- morphism, co‐dominant inheritance, and high reproducibi-lity (Chistiakov., 2006; Song., 2017; Liu.,2019). In addition, SSR primers can be transferred from one species to another, thus greatly reducing the need for com-plex primer development in related species (Song.,2017). To explore the genetic characteristics of,Xu. (2011) detected 21 specific SSR markers using an enriched library. However, the low efficiency and polymor-phism have limited further investigations (Zhu., 2014).
With the advent of high-throughput sequencing techno- logies,SSR markers underwent large-scale development for a variety of marine non-model species (Li., 2019).These new methods have revolutionarily ushered in an op-portunity for developing the highly polymorphic SSR mar- kers of. In this study, 26 polymorphic SSR mar- kers ofwere screened. Cross-species transfer- ability was tested in three related species which belonging to the same suborder Myctophoidei. This work will provide powerful tools for further population genetic studies onand assist the conservation of its germplasm re- sources.
2 Materials and Methods
2.1 Sample Collection and Genomic DNA Extraction
Oneindividual was captured from the coas- tal water of Zhoushan, China, during September 2020, for high-throughput sequencing. The other 24 individuals werecollected from four geographical locations, namely, Zhou- shan, Qingdao, Beihai, and Sanya, and used for primers’ poly-morphism detection (Table 1). Thirty individuals of related species, including,, and, were employed for cross- species transferability examination. Fresh muscle tissues were fixed in RNAhold (TransGen Biotech, Beijing, China) solution or absolute ethanol and then stored at −80℃ in theFisheries Ecology and Biodiversity Laboratory of Zhejiang Ocean University, Zhoushan, China,prior to further research.DNA was extracted following the traditional standard phe- nol-chloroform protocol. The purity, concentration, and in- tegrity of RNA were checked by Nanodrop (ThermoFisher, Massachusetts, USA), Qubit (Invitrogen, California, USA), and Agilent 2100 (Agilent, California, USA). The degrada- tion and pollution of extracted DNA were detected by 1% agarose gel electrophoresis.

Table 1 Fish material collected for this study
2.2 High-Throughput Sequencing and SSR Identification
The RNA sample was frozen on dry ice and sent to San-gon Biotech (Shanghai) Co., Ltd., China for cDNA library construction andpaired-end transcriptome sequencing with150bp on Illumina HiseqTM2500 platform. Fast QC (An-drews, 2010) and Trimmomatic software (Bolger., 2014) were primarily used to evaluate the quality of raw data andobtain clean data, respectively.sequence assembly was then performed using Trinity software (Grabherr., 2011). The longest transcript in each transcript cluster was selected as the representative unigene and used for micro- satellite identification with the software Microsatellite (Beier., 2017). The standards of screening SSR include re- peat motif length (1–6bp), the minimum threshold of re- peat counts (motif type-repeats: 1-10, 2-6, 3-5, 4-5, 5-5, and 6-5), and identification ofcompound SSRs (interrup- tion between two SSRs was less than 100bp).
2.3 Primer Development and Polymorphism Detection
A total of 100primer pairs were designed using the Pri- mer3 version 2.37 (http://bioinfo.ut.ee/primer3-0.4.0/) un- der the following parameter settings: GC content ranged from 40% to 60%, repeat counts of motifs were more than 6, the expected size of PCR product was adjusted to 150–300bp, and polyadenylation should be avoided. The primers were synthesized by Shanghai Sunny Biotechnology Co., Ltd., China, and then were tested on the 24 individuals sam- pled for the polymorphism detection. PCR amplification was performed in a 25μL volume reaction system contain- ed 2.5μL of 10× PCR buffer (Trans, Beijing), 2μL of dNTPs, 1μL each of forward and reverse primers (10μmolL−1), 17.25μL of deionized water, 0.25μL of Easy Taq DNAPo- lymerase (Trans, Beijing), and 1μL of DNA template. PCR conditions were as follows: initial denaturation (94℃ for 5min), followed by 33 cycles of denaturing (94℃ for 30s), annealing (30s at optimum annealing temperature) and elongation (30s at 72℃), and an additional extension (72℃for 10min). The amplification products of each specific pri- mer were detected by 8% non-denaturing polyacrylamide gel electrophoresis and visualized by silver staining.
2.4 Cross-Species Amplification
Transferability was assessed by PCR amplification to test the cross-species amplification of the developed SSR loci, and polyacrylamide gel electrophoresis was conducted to detect the abovementioned product on 10 samples of each related species. PCR amplification was performed as de- scribed above.
2.5 Data Analysis
The genetic parameters of the number of alleles (a), ob- served heterozygosity (o), expected heterozygosity (e), and the polymorphism information content () were as- sessed using Excel microsatellite toolkit version 3.1 (Shai- bi., 2008). The presence of null alleles was inspected by Micro-Checker (Oosterhout., 2004). Genepop 4.0 was employed to analyze Hardy-Weinberg equilibrium (Rousset, 2008).
3 Results and Discussion
3.1 Raw Data’s Quality
Illumina-based RNA sequencing for muscle tissue’s cDNAlibrary resulted in 41886302 raw reads and 39328676 cleanreads after filtering, thus corresponding to 5584743187 nu- cleotides. The average Q20, Q30, and GC percentages were97.94%, 91.58%, and 51.39%, respectively. A total of 35859 contigs were assembled and clustered into 29756 unigenes with an N50 length of 797bp. The results of assembly ana- lysis are displayed in Table 2. Overall, the high-quality tran- scriptomic sequencing and assembly results indicated that the data could be used for further analysis.

Table 2 Statistical results of transcriptome data assembly
Notes: N50 represents a weighted median statistic that 50% of the total length is contained in unigenes greater than or equal to this va- lue. N90 represents a weighted median statistic that 90% of the total length is contained in unigenes greater than or equal to this value.
3.2 Characterization of SSRs in the H. nehereus Transcriptome
A total of 5652 potential SSRs (excluded 551 compoundSSRs) were identified in 16974320 unigenes; That is, 332.97SSR loci were found per Mb of the unigenes. Compared with those of other commercial or ornamental fishes such as(153.00 SSRMb−1) (Huang., 2019),(429.46 SSRMb−1) (Zhao., 2019), and(100.51 SSRMb−1) (Wang., 2016), the SSRs oftranscriptome were moderate. Among all these SSRs, the mononucleotide motif (67.59%) was the most abundant, followed by dinu- cleotides (20.72%) and trinucleotides (8.97%). Tetranucleo- tide, pentanucleotide, and hexanucleotide showed a relative- ly low proportion at only 2.72% in total (Fig.1). Mononu- cleotide repeats are common in most eukaryotic genomes (Wierdl., 1997) and might be relevant to the frequent mutation and poor stability of long microsatellite sequences(Song., 2019). Furthermore, the abundance of low- le- vel repeat motifs indicated thathad reached a high evolution level and had a wide variation frequency and greatpotential for polymorphic marker development (Li., 2017; Harr and Schlötterer, 2000).

Fig.1 Distribution of different repeat units in H. nehereus.
The results of motif type detection showed that within the mononucleotide repeats, A and T motifs had a high propor- tion that was approximately 27 times that of G and C mo- tifs (Fig.2a). In particular, the A and T repeats contributed65.20% of the total numbers of SSRs. In dinucleotides, GT(16.74%), TG (16.57%), and AC (16.23%) repeat SSRs weredominant (Fig.2b). CG repeats were rarely found, which isin accordance with the conclusions for(Sun., 2019),(Wang., 2007),(Takagi., 2003), and many other fishes. Schotterer. (1992) considered that the above phenomenon is mainly caused by CpG methylation in genomic-making that allows C to transform to T through DNA deamination. In trinucleotide repeats, GAG (5.72%), GAT (4.93%), and TTG (4.54%) repeat motifs were the most common (Fig.2c). In addition, tetranucleotide repeats had a high frequency of TTTG and AAAC repeat types and pre-sented the same feature of abundant A and T contents as that of mononucleotide repeats (Fig.2d). In pentanucleotide and hexanucleotide repeat units, each motif type was even-ly distributed, and no advantaged repeat type was observed (Figs.2e, f). The copy number of repeat motifs ranged from 5 to 58, and 10 repeat units (23.23%) were the most repre- sented types (Fig.3).
3.3 Polymorphism Detection of SSR Primers
A total of 100 primer pairs were randomly selected and primarily amplified in eightspecimens by gra- dient temperature PCR to determine the optimum anneal- ing temperature of each primer. Eventually, 80% (80 loci) were successfully amplified. Imperfections in transcripto- mic assembly and the presence of introns between primers are the possible causes of the failed amplification (Wang., 2019). The validated markers were subsequently am- plified in 24 individuals sampled from four different loca- tions. A representative electrophoretogram (LTY-75) forpolymorphic locus is shown in Fig.4. The results showed 26 pairs of primers that were polymorphic and accounted for 32.5% of the total effective amplification primers. These lociconsisted of 24 dinucleotide repeat motifs, 1 trinucleotide,and 2 tetranucleotides. Because of their abundant distribu-tion in the vertebrate genome, dinucleotide SSR markers are widely applied in molecular ecology (Liu, 2017). Mean-while, effective and visually genetic parameters are still need- ed for the assessment of suitable molecular markers. 3 to 11 at each locus) with an effective average of 6.89±1.83. As a significant index of genetic variation reflecting genetic diversity and structure stability, heterozygosity is often used to assess SSR primers’ amplification effect on the selected stock (Wang., 2019; Song., 2020). In this study, the expected (e) and observed (o) hetero- zygosities were 0.355–0.885 with a mean value of 0.761±0.115, and 0.375–0.958 with a mean value of 0.753±0.166, respectively. Except for LTY-42, all of the loci had rela- tively high heterozygosity. The polymorphic information content is a crucial indicator of the polymorphisms of gene- tic markers. Theof the 26 loci ranged from 0.316 to 0.852 with a mean value of 0.713±0.120. Botstein. (1980) reported that except for two moderate polymorphic loci (LTY-42 and LTY-83), the remaining 24 loci all dis- played high polymorphism (>0.5). Compared with the polymorphic primers identified by the magnetic bead en- richment method (the mean values ofa,e,oandwere 3.6, 0.592, 0.558, and 0.521, respectively) (Xu., 2011),the present results revealed high efficiency and poly-morphism. We inferred that the reasons lie in the variation of population genetic diversity under different time scales, the selection of repeat types, and the progress of sequenc- ing technology. In addition, the collection of specimens from multiple sample sites may be an underlying cause of high polymorphism (Zheng., 2020).
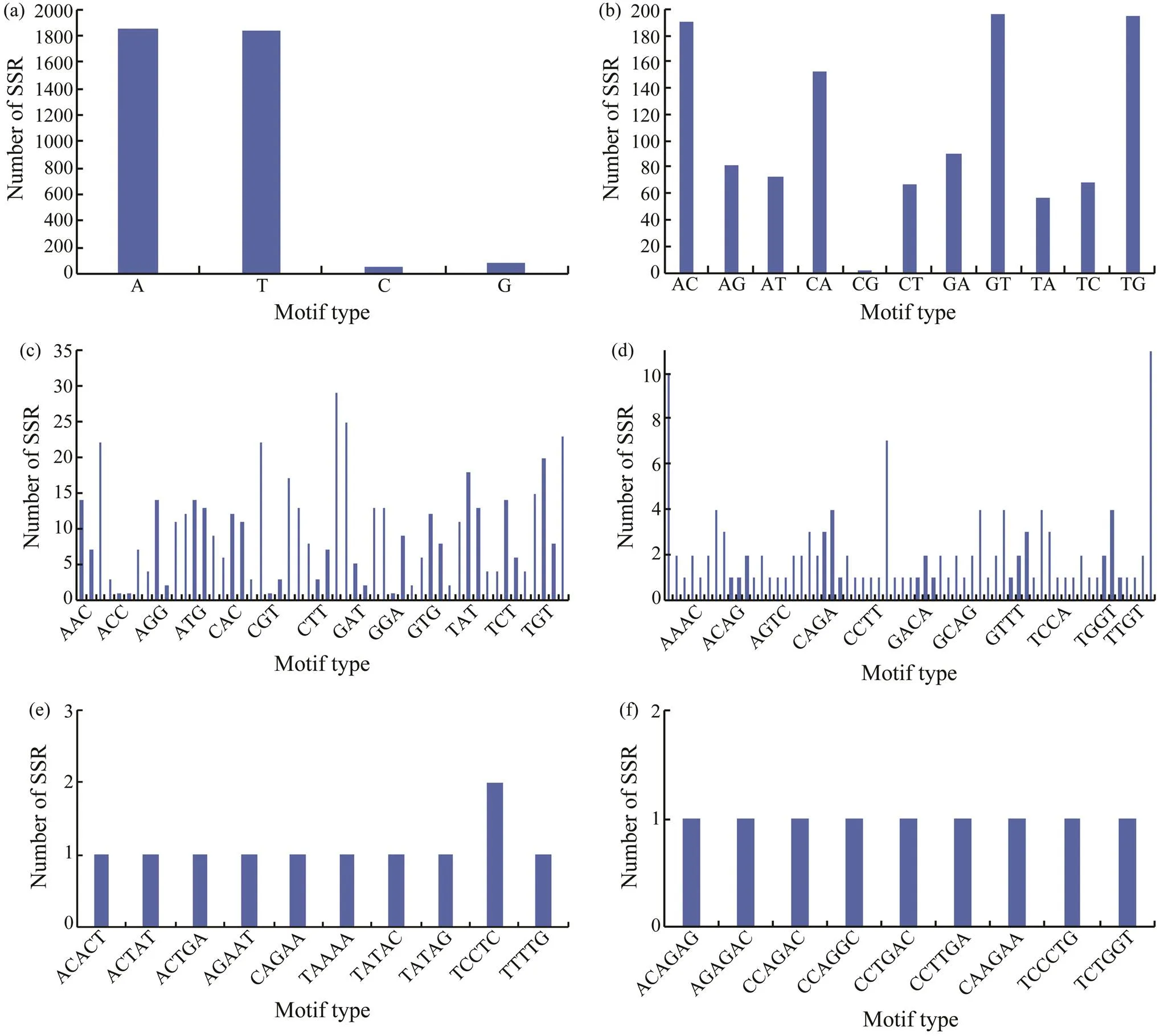
Fig.2 Characterization and number of different motifs among mononucleotide(a), dinucleotide(b), trinucleotide(c), te- tranucleotide(d), pentanucleotide(e) and hexanucleotide(f).

Fig.3 Repeats counts and number of SSR for different motif types.

Fig.4 Intraspecies diversity among 24 samples identified by the locus of LTY-75.Bands with different lengths indicate different alleles; M represents DNA Marker.
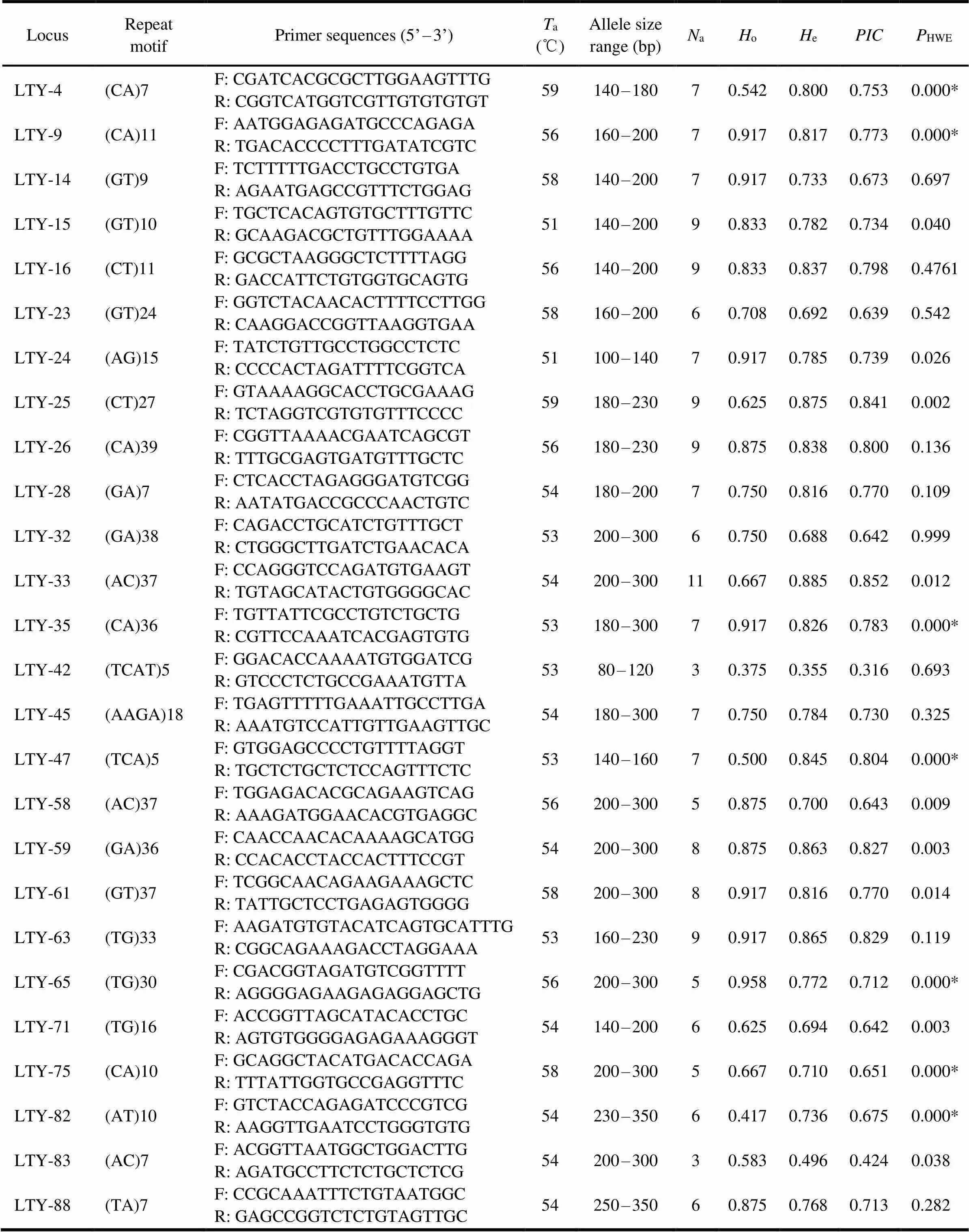
Table 3 shows the 179 detected alleles (Na) (ranging fromTable 3 Characterization of 26 SSRs for H. nehereus
Notes:a, optimized annealing temperature;a, number of alleles observed;o, observed heterozygosity;e, expected heterozygosity;, polymorphism information content. *Significant deviation from HWE after Bonferroni correction (<0.0019).
Hardy-Weinberg equilibrium (HWE) testing revealed se- ven polymorphic loci that significantly deviated from equi-librium after sequential Bonferroni correction (<0.0019).Furthermore, null alleles and significant differences betweeneandowere detected from the majority of the seven loci.owas remarkably lower thane, a phenomenon con-comitant to the existence of null alleles and indicates thepoor stability of gene and genotype frequency (Qi., 2013; Park., 2020). We inferred that the presence of null alleles may be an important factor for the deviation from HWE. In general, the 26 microsatellite markers with highpolymorphism and genetic variation in this study were app- licable to the further study on the population genetics of.
3.4 Cross-Species Amplification of Polymorphic SSR Loci
Microsatellite is one of the most commonly used gene- tic markers in the study of population genetics; however,the lack of ‘universal’ PCR primers increases the difficulty of related research to a great extent (Primmer., 2005). A growing number of studies recently showed that cross-species microsatellite amplification in related species is aneffective and viable strategy. However, in the present work, the amplification of the three related species was unsatis- factory. Only two loci (LTY-45 and LTY-65) showed spe-cific amplification inand, respec- tively. Other loci similar to LTY-59, LTY-75, and LTY-4 could be amplified successfully but exhibit unclear electro- phoresis bands. The transferability of SSR markers is direct-ly related to the evolutionary divergence between the sourcespecies, from which the SSR loci have been isolated, andthe target species, in which the heterologous loci are be- ing applied (Primmer., 2005). Molecular phylogenic analysis based on mitochondrial Cytgene showed that the genetic distance amongand related genera was mainly 0.215–0.350, which is farther than that from other cross-species amplification research (Liang., 2019; Yang., 2020). If different species from the same genus are selected, the primers’ transferability may increase. Un-fortunately, only one closest relative of() has been reported in the coastal waters of China. Given thatis a rare species and only has a narrow distribution in the Taiwan Strait, sample col-lection is difficult. Considering the limited transferability of SSR markers at the transcriptome level and the scarcity of the research target in this study, we hope to collect addi- tional related species and excavate SSRs using genome- based technologies to save and improve the inadequacies in the follow-up work.
4 Conclusions
This study analyzed the characteristics of microsatellitesfromusing high-throughput sequencing. Twen- ty-six polymorphic SSR markers were developed and test- ed for their cross-species amplification abilities. These no-vel markers can be applied in the population genetic diver- sity research ofand provide a useful reference for resource conservation and utilization.
Acknowledgements
This research was supported by the Scientific Research Projects of Department of Education of Zhejiang Province (No. Y201942611), the Science and Technology Innova- tion Project of College Students in Zhejiang Province (No. 2021R411008), and the Open Foundation from Marine Sci-ences in the First-Class Subjects of Zhejiang Province.
Andrews, S., 2010. FastQC a quality control tool for high through-put sequence data. Availableat: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M., 2017. MISA-web: A web server for microsatellite prediction., 33 (16): 2583, DOI:10.1093/bioinformatics/btx198.
Bolger, A. M., Lohse, M., and Usadel, B., 2014. Trimmomatic: A flexible trimmer for Illumina sequence data., 30 (15): 2114-2120, DOI:10.1093/bioinformatics/btu170.
Botstein, D., White, R. L., Skolnick, M., and Davis, R. W., 1980. Construction of a genetic linkage map in man using restrictionfragment length polymorphisms., 32 (3): 314-331.
Chakraborty, P., Sahoo, S., Bhattacharyya, D. K., and Ghosh, M., 2020. Marine lizardfish () meal concentrate in preparation of ready-to-eat protein and calcium rich extruded snacks., 57 (1): 338- 349, DOI:10.1007/s13197-019-04066-0.
Chen, S. Z., 2002.. Science Press, Beijing, 27-85 (in Chinese).
Chistiakov, D. A., Hellemans, B., and Volckaert, F. A. M., 2006. Microsatellites and their genomic distribution, evolution, func- tion and applications: A review with special reference to fishgenetics., 255 (1-4): 1-29, DOI: 10.1016/j.aquacul ture.2005.11.031.
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I.,., 2011. Full-length transcriptome assem- bly from RNA-Seq data without a reference genome., 29 (7): 644-652, DOI: 10.1038/nbt.1883.
Guo, J. H., Li, J., Shen, C., Shi, Y., Feng, C., He, X. B.,., 2019a. Estimation of biological parameters and stock ofin the Min River Estuary, East China Sea., 39 (5): 56-64, DOI: 10. 3969/j.issn.1673-9159.2019.05.009 (in Chinese with English abstract).
Guo, Y. J., Yang, T. Y., Meng, W., Han, Z. Q., and Gao, T. X., 2019b. The genetic structure of the Bombay Duck () based on mitochondrial Cytgene., 43 (5): 945-952, DOI: 10.7541/2019.112 (in Chi- nese with English abstract).
Harr, B., and Schlötterer, C., 2000. Long microsatellite alleles inhave a downward mutation bias and short persistence times, which cause their genome-wide under-representation., 155 (3): 1213-1220, DOI: 10.1093/ genetics/155.3.1213.
Hasin, T., 2016. Length-weight relationship and condition factor of bombay duckfrom landings of fishery ghat, Chittagong., 3 (2): 355-360, DOI: 10.3329/ralf.v3i2.29367.
He, X. B., Li, J., Shen, C., Shi, Y., Feng, C., Guo, J. H.,., 2018. Length-weight relationship and population dynamics of Bombay duck () in the Min River Estuary, East China Sea., 35: 253-261, DOI: 10.1007/s41208-018-0117-7.
Hedrick, P. W., 2001. Conservation genetics: Where are we now?, 16 (11): 629-636, DOI: 10. 1016/S0169-5347(01)02282-0.
Huang, Y., Gong, W. B., Chen, H. G., Xiong, J. L., and Sun, X. H., 2019. Sequencing and bioinformatic analysis for transcriptome ofbased on RNA-seq., 15 (1): 106-112, DOI: 10.12131/20180066 (in Chinese with English abstract).
Jang, W., Jang, Y., Kim, N. H., Waminal, N. E., Kim, Y. C., Lee, J. W.,., 2020. Genetic diversity among cultivated and wildpopulations revealed by high-resolution micro-satellite markers., 44 (4): 637- 643, DOI: 10.1016/j.jgr.2019.05.008.
Jiang, Y. L., Meng, W., Zhu, W. B., and Yang, T. Y., 2020. Gene- tic diversity analysis ofpopulations based on mitochondrial DNAgene., 39 (2): 102-109 (in Chi- nese with English abstract).
Li, C. J., Teng, T., Shen, F. F., Guo, J. Q., Chen, Y. M., Zhu, C. K.,., 2019. Transcriptome characterization and SSR disco- very in., 37 (1): 235-244, DOI: 10.1007/s00343-019-7298-7.
Li, T. Q., Liu, X. F., Wan, Y. M., Li, Z. H., Li, Y. Y., Liu, X. X.,., 2017. Characteristic analysis of microsatellites in the tran-scriptome of, an endangered species endemic to southeastern Yunnan, China., 30 (4): 533-541, DOI: 10.13275/j.cnki.Lykxyj.2017.04. 001 (in Chinese with English abstract).
Liang, Z. B., Wu, R. X., Zhu, S. F., and Luo, H. G., 2019. Micro- satellite loci development inby SLAF-seq technology and cross-amplification in five Carangidae fish- es., 39 (4): 5-12, DOI: 10.3969/j.issn.1673-9159.2019.04.002 (in Chinese with Eng- lish abstract).
Liu, B. J., 2017. Population genetic structure and local adaptationof the small yellow croaker () and Japa-nese eel (). PhD thesis. Chinese Academy of Sciences (in Chinese).
Liu, Q., Liu, L., Song, N., Wang, X. H., and Gao, T. X., 2019. Ge- netic structure in the marbled rockfish () across most of the distribution in the northwestern Pacific., 35: 1249-1259, DOI: 10.1111/ jai.13972.
Lu, G., Basley, D. J., and Bernatchez, L., 2001. Contrasting pat- terns of mitochondrial DNA and microsatellite introgressive hybridization between lineages of lake whitefish (); relevance for speciation., 10(4): 965-985, DOI: 10.1046/j.1365-294x.2001.01252.x.
Nugroho, D., Faizah, R., Prasetyo, A. P., and Badrudin, M., 2015. Bio-exploitation status of Bombay duck (Hamilton, 1822) on trawl fishery in Tarakan waters., 21 (1): 53-59, DOI: 10.15578/ifrj. 21.1.2015.53-59.
Oosterhout, C. V., Hutchinson, W. F., Wills, D. P. M., and Shipley P., 2004. Micro-checker: Software for identifying and correct- ing genotyping errors in microsatellite data., 4 (3): 535-538, DOI: 10.1111/j.1471-8286.2004.00684.x.
Park, Y. J., Lee, M. N., Kim, E. M., Park, J. Y., Noh, J. K., Choi, T.J.,., 2020. Development and characterization of novel polymorphic microsatellite markers for the Korean freshwater snailand cross-species amplification using next-generation sequencing., 38 (2): 503-508, DOI: 10.1007/s00343-019-9058-0.
Primmer, C. R., Painter, J. N., Koskinen, M. T., Palo, J. U., and Merilä, J., 2005. Factors affecting avian cross-species micro- satellite amplification., 36 (4): 348- 360, DOI: 10.1111/j.0908-8857.2005.03465.x.
Qi, X. Y., Dong, Y. H., Yao, H. H., Zhou, X. L., and Lin, Z. H., 2013. Development of 30 microsatellite markers inand their application inand., 37 (8): 1147-1154, DOI: 10.3724/SP.J.1231.2013.38484 (in Chinese with English abstract).
Rousset, F., 2008. Genepop'007: A complete re-implementation of the genepop software for Windows and Linux., 8 (1): 103-106, DOI: 10.1111/j.1471-8286. 2007.01931.x.
Sarker, M. N., Humayun, M., Rahman, M. A., and Uddin, M. S., 2017. Population dynamics of Bombay duck(Hamilton, 1822) of the Bay of Bengal along Bangladesh coast., 45 (2): 101-110, DOI: 10.3329/bjz.v45i2.35705.
Schorderet, D. F., and Gartler, S. M., 1992. Analysis of CpG sup- pression in methylated and nonmethylated species., 89 (3): 957-961, DOI: 10. 1073/pnas.89.3.957.
Shaibi, T., Lattorff, H., and Moritz, R., 2008. A microsatellite DNAtoolkit for studying population structure in., 8 (5): 1034-1036, DOI: 10.1111/j. 1755-0998.2008.02146.x.
Song, C. Y., Feng, Z. Y., Li, C. H., Sun, Z. C., Gao, T. X., Song, N.,., 2020. Profile and development of microsatellite pri- mers forbased on high-throughput sequencing technology.,38 (6): 1880-1890, DOI: 10.1007/s00343-019-9154-1.
Song, N., Chen, M. Y., Gao, T. X., and Yanagimoto, T., 2017. Pro- file of candidate microsatellite markers inusing 454 pyrosequencing., 35 (1): 198-202, DOI: 10.1007/s00343-016- 5103-4.
Song, Q., Guo, X. G., and Chen, D. L., 2019. Characterization of microsatellite DNA loci and design of candidate primers toamplify these regions forby using454 GS FLX., 38 (5): 512-520,DOI: 10.11984/j.issn.1000-7083.20190010 (in Chinese with English abstract).
Sun, H. L., Sun, C. F., Dong, J. J., Tian, Y. Y., Hu, J., and Ye, X., 2019. Transcriptome sequencing and development and applica- tion of novel SSR markers for., 38 (10): 4413-4421, DOI: 10.13417/j.gab.038. 004413 (in Chinese with English abstract).
Takagi, M., Sato, J., Monbayashi, C., Aoki, K., Tsuji, T., Hatana- ka, H.,., 2003. Evaluation of microsatellites identified in the tiger pufferDNA database., 69 (6): 1085-1095, DOI: 10.1111/j.0919-9268.2003.00 732.x.
Taqwa, A., Burhanuddin, A. I., Niartiningsih, A., and Nessa, M. N., 2020. Nomei fish (, Ham. 1822) repro-duction biology in Tarakan waters., 473:012012, DOI: 10.1088/ 1755-1315/473/1/012012.
Wang, D., Liao, X. L., Cheng, L., Yu, X. M., and Tong, J. G., 2007. Development of novel EST-SSR markers in common carp by data mining from public EST sequences., 271 (1-4): 558-574, DOI: 10.1016/j.aquaculture.2007.06.001.
Wang, Q. L., Liu, Y., Song, H. M., Wang, X. J., Liu, C., Mu, X. D.,., 2016. Development and primer selection of EST-SSR molecular markers based on transcriptome sequencing of., 46 (6): 8-13, DOI:10.13721/j.cnki.dsyy.2016.06.002 (in Chinese with Eng- lish abstract).
Wang, Z. M., Li, J. H., Hao, R. J., Adzigblia, L., and Deng, Y. W., 2019. Characterization and development of SSR markers ofby RNA-Seq approach., 15: 2352-5134, DOI: 10.1016/j.aqrep.2019.100230.
Wierdl, M., Dominska, M., and Petes, T. D., 1997. Microsatellite instability in yeast: Dependence on the length of the microsa- tellite., 146 (3): 769-779, DOI: 10.1093/genetics/146. 3.769.
Xu, T. J., Sun, D. Q., Li, H. Y., and Wang, R. X., 2011. Develop- ment and characterization of microsatellite markers for the li- zardfish known as the Bombay duck,(Sy- nodontidae)., 10 (3): 1701- 1706, DOI: 10.4238/vol10-3gmr1045.
Yang, T. Y., Jiang, Y. L., Guo, Y. J., Zhu, L. Q., Lin, Y. J., Zheng, Y. J.,., 2020. Molecular phylogeny ofand its close relatives based on mitochondrial Cytgene., 6: 77-85, DOI: 10. 13984/j.cnki.cn37-1141.2020.06.010 (in Chinese with English abstract).
Zane, L., Bargelloni, L., and Patarnello, T., 2002. Strategies for microsatellite isolation: A review., 11 (1): 1-16, DOI: 10.1046/j.0962-1083.2001.01418.x.
Zhang, B., and Jin, X. S., 2014. Feeding habits and ontogenetic diet shifts of Bombay duck,., 32 (3): 524-548, DOI: 10. 1007/s00343-014-3085-7.
Zhang, B., Tang, Q. S., and Jin, X. S., 2009. Functional groups of communities and their major species at high trophic level in the Yellow Sea ecosystem., 29 (3): 1009- 1111 (in Chinese with English abstract).
Zhao, Y. H., Qu, Y. J., Wen, J. F., Li, J. E., and Zhou, H., 2019. Development of SSR markers inby tran- scriptome sequencing., 50 (9): 2078-2087, DOI: 10.3969/j.issn.2095-1191.2019.09.26 (in Chi- nese with English abstract).
Zheng, J., Song, C. Y., Gao, T. X., and Song, N., 2020. Profile and development of microsatellite markers for the small yellow croakerbased on High-throughput Se- quencing Technology., 38: 2352-4855, DOI: 10.1016/j.rsma.2020.101370.
Zhu, Z. H., Li, H. Y., Qin, Y., and Wang, R. X., 2014. Genetic di- versity and population structure inbased on sequence-related amplified polymorphism markers., 13 (3): 5974-5981, DOI: 10.4238/ 2014.august.7.13.
(May 27, 2021; revised August 3, 2021; accepted April 26, 2022)
© Ocean University of China, Science Press and Springer-Verlag GmbH Germany 2023
Corresponding author. E-mail: hellojelly1130@163.com
(Edited by Qiu Yantao)
杂志排行

Journal of Ocean University of China的其它文章
- The Subduction Structure Beneath the New Britain Island Arc and the Adjacent Region from Double-Difference Tomography
- Differences of Polygonal Faults with Irregularly Polygonal Geometries: A Case Study from the Changchang Sag of Qiongdongnan Basin, Northern South China Sea
- Characterization of Bacterial Communities in Aerosols over Northern Chinese Marginal Seas and the Northwestern Pacific Ocean in Autumn
- Assessment and Application of Beach Quality Based on Analytic Hierarchy Process in Yangkou Beach, Qingdao
- Role of Resuspended Sediments as Sources of Dissolved Inorganic Phosphorus Along Different Dimensions in the Subei Shoal, South Yellow Sea, China
- Pharmacokinetics of Enrofloxacin and Its Metabolite in Carp (Cyprinus carpio) After a Single Oral Administration in Medicated Feed