基于GPU 的固态晶体硅分子动力学算法优化
2023-03-16祝爱琦侯超峰
李 靖,祝爱琦,韩 林,侯超峰
(1.郑州大学 信息工程学院,郑州 450001;2.中国科学院过程工程研究所,北京 100190;3.郑州大学 国家超级计算郑州中心,郑州 450001)
0 概述
近年来,利用图形处理器(Graphics Processing Unit,GPU)加速分子动力学(Molecular Dynamics,MD)模拟已成为计算化学、计算材料学等领域的研究热点[1-2]。分子动力学模拟通常从原子、分子尺度上研究凝聚态材料的热力学性质和行为,被广泛应用于微观计算模拟领域中[3]。晶体硅是信息技术产业中应用最广泛和最重要的材料之一,其纳微结构的热传递性质和导热性能会显著影响电子设备的稳定性,导致器件性能降低。因此,分子动力学模拟方法对接近真实尺度硅纳微结构的直接模拟的实现与其导热机制的研究具有重要意义。
模拟体系的计算量和时空分辨率的提高是分子动力学模拟领域的研究热点。高性能加速芯片(如GPU)为处理模拟体系的巨量计算提供契机。GPU作为一种具有良好并行性能且适应密集计算的新型加速计算工具,在通用科学计算领域具有较大的应用潜力。分子动力学模拟优化的研究大多集中在通用分子动力学模拟算法优化和程序并行算法设计方面,很少专门针对固体材料并结合GPU 硬件结构特性对MD 算法进行设计和优化。文献[4]提出一种高效的多粒子碰撞动力学算法,并在多个GPU 上实现。针对多体势的高效分子动力学模拟,文献[5]提出一种基于GPU 的力求解算法。文献[6]实现分子动力学三体势的GPU 加速计算。文献[7]提出不同于对势分子动力学模拟的方法,根据复杂的多体势描述一种GPU 加速的固态共价晶体分子动力学模拟的高效和可扩展算法。
针对固态共价晶体硅的分子动力学模拟,本文结合原子间多体势函数与CUDA 编程模型,通过对GPU 并行计算特征和模拟热点进行分析,提出面向GPU 计算平台的固定邻居算法设计与优化。根据GPU 硬件架构特性,将并行线程处理的float4 数据结构重新映射为连续float 布置的形式,采用三目运算符控制指令排队,消除条件分支语句造成的流水线停顿,减少全局访存消耗和分支结构耗时,提升算法的计算效率。
1 Tesla V100 GPU 硬件架构
为充分发挥GPU 的计算性能,从软件与硬件相结合的角度分析固态材料分子动力学模拟算法。以Tesla V100 GPU 为实验平台描述GPU 硬件架构[8-10]。Tesla V100 GPU 架构如图1 所示,由多个图形处理簇(Graphics Processing Cluster,GPC)、纹理处理簇(Texture Processing Cluster,TPC)、流式多处理器(Streaming Multiprocessor,SM)以及内存控制器组成。每个SM 内有64 个流处理器(Streaming Processor,SP),其中,每个SP 具有自己的缓存空间和寄存器组,并通过线程束(warp)调度器的方式控制SP 的并行执行,将L1 高速缓存和共享内存组合到单个内存块中,内存总容量为128 KB,在SM 上运行的线程对L1 高速缓存和共享内存的访问相对于全局内存具有更快的访问速度,在不同SM 之间的L1 高速数据缓存和共享内存相互独立。Tesla V100 GPU 总共拥有80 个SM、5 120 个32 位浮点处理单元、2 560 个64 位浮点处理单元、640 个Tensor 核心以及320 个纹理单元(Tex)[8]。
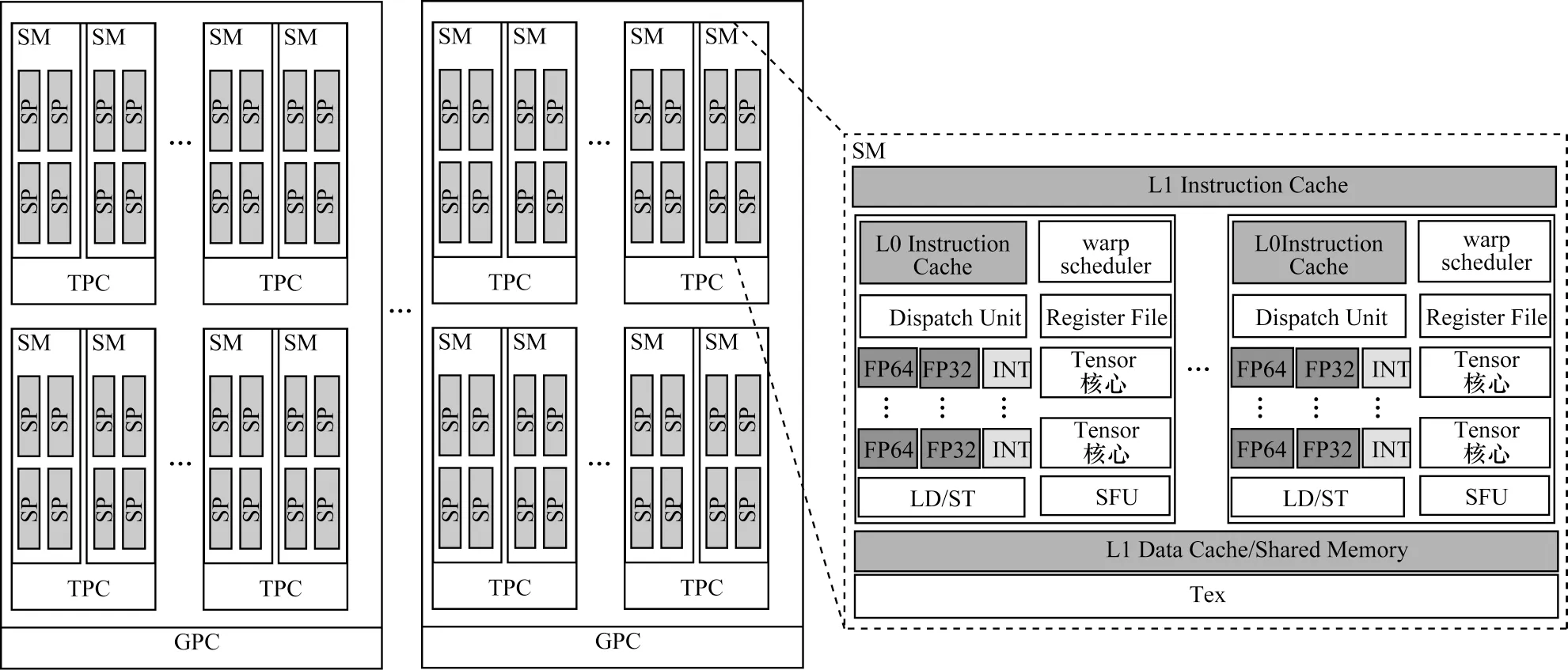
图1 Tesla V100 GPU 架构Fig.1 Architecture of Tesla V100 GPU
在硬件与执行程序的对应关系上,一个SP 控制一个线程的执行。为提高并行效率,NVIDIA 把32 个线程组织成线程束的形式,并在warp 内以单指令多线程(Single Instruction Multiple Threads,SIMT)的方式执行。在线程块中的多个warp 由线程束调度器调度进入SM 执行[11]。在Tesla V100 GPU 中大量的SM 虽然提高GPU 硬件的并行性,但是其具有较高并行性的复杂架构给MD 模拟算法的优化带来挑战。在线程并行优化方面,不仅减少线程之间的相互依赖关系,还需要充分利用其硬件特性以减少访存开销。
2 固定邻居算法在GPU 上的多体势实现
2.1 Tersoff 多体势的分子动力学模拟
MD 模拟是通过刻画微观原子的运动过程,由相关的数据统计来研究模拟体系的结构和热力学性质的方法。其中,在周围邻居原子势场作用下每个原子根据牛顿定律运动[12]。原子间的相互作用是MD 模拟算法的主要组成部分。与原子间对势相比,原子间多体势具有更复杂的模型形式,但通常能更准确、更有效地体现共价材料模拟体系的物理特性[13]。
本文针对固态共价晶体硅,基于固定邻居算法,利用Tersoff 多体势实现晶体硅的MD 模拟计算。Tersoff 势函数的数学表达式如式(1)和式(2)所示:
其中:UR为排斥势项;UA为吸引势项;fC为截断函数;bij为键级项。UR、UA、fC、bij、ζij和g(θ)的数学表达式如式(3)~式(8)所示:
其中:ζij为原子i与原子j间的角势能。上述公式的模型参数见文献[14-15]。固态共价晶体硅的MD 模拟计算首先需要生成模拟体系晶体硅原子的初始位置,并在特定温度下给出原子的初始速度,通过Tersoff 多体势计算每个硅原子受其他硅原子的作用合力,然后选择蛙跳算法积分牛顿运动方程,求解硅原子的新速度和新位置。
2.2 固定邻居算法的GPU 实现
固态共价晶体硅具有规则的晶胞结构,在低温状态下每个原子周围有4 个邻居原子。在Tesla V100 GPU 平台实现算法时,模拟体系中所有的硅原子按超胞[7]的顺序在分配的设备存储器上排列,并且在后续的迭代模拟中按超胞顺序分配的内存位置保持不变。超胞中原子的分组情况如图2 所示。根据每个原子的邻居原子在超胞内的数目,把超胞内的原子分为三组:第一组原子在超胞内有一个邻居原子,如图2(a)中的灰色原子;第二组原子在超胞内有两个邻居原子;第三组原子在超胞内有四个邻居原子,如图2(a)中的黑色原子。算法执行时将超胞内上述三组原子按顺序布置,如图2(b)所示。此外,为提高缓存效率,采用静态重排方法代替动态更新邻居列表的过程,根据超胞内原子分组情况,把原子在超胞内和超胞外的邻居原子信息分组聚合并进行连续排列。由于固定邻居算法没有邻居列表动态更新的过程,因此原子间作用力的计算、原子位置和速度的更新占大量的模拟总时间。
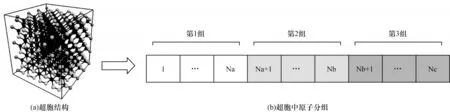
图2 每个超胞中计算原子的分组示意图Fig.2 Schematic diagram of grouping of calculated atoms in each supercell
固定邻居算法流程如图3 所示,在CPU 端通过控制模拟的原子数目和迭代步数,初始化原子的位置、速度等信息,将原子按其下标映射到与超胞排序相应的CPU 端内存数组中,然后完成CPU 到GPU 的数据拷贝以及GPU 端核函数的生成和调度。在GPU 端,首先将原子按其下标映射到超胞排序的相应GPU 数组中,根据“一个线程模拟一个原子”的分配原则,使超胞中的原子数等于线程块中的线程数,并利用线程分支控制超胞内的分组情况。在GPU设备端并行计算每个超胞中每个原子和其邻居原子之间的距离,进一步计算每个原子受邻居原子的全部作用力。最后,通过积分时间计算原子的新速度和新位置。当算法没有达到模拟设定的迭代步数时,GPU 端继续执行MD 模拟,直到达到设定的模拟迭代步数,程序结束。

图3 固定邻居算法流程Fig.3 Procedure of fixed neighbor algorithm
虽然固定邻居算法采用超胞分组的方式提高缓存命中率,但是在设备端并行执行原子受力计算时也增加了if 分支,为GPU 程序并行执行带来了额外的分支消耗。Tersoff 多体势比对势具有更复杂的模型形式,当计算原子间作用力时使用数据类型(float4)并未实现计算吞吐量和数据全局访存的最佳匹配。因此,为提高程序的性能,本文结合GPU硬件架构特性,对固定邻居算法进行设计优化。
3 固定邻居算法优化实现
在对Tesla V100 GPU 平台的硬件特性进行分析之后,本文分别对固定邻居算法的数据结构和分支结构进行优化,充分利用GPU 计算资源来提高线程并行效率,进一步提升固定邻居算法的性能。
3.1 数据结构优化
早期的GPU 适用于图形渲染设计,其运算对象是表示颜色的R、G、B、A 和表示坐标的X、Y、Z、W,都是对一个四维向量进行操作[16]。随着GPU 应用越来越广泛,数据结构可能不仅具有表示坐标的X、Y、Z、W和表示颜色的R、G、B、A,还有可能存在一些非向量的数据。因此,本文对不同的数据做不同的处理,而不同的数据结构方式会影响数据的内存局部性。为提高通用性,NVIDIA 引入标量计算的概念,当发展到Tesla V100 时,其内部包含大量专门针对标量的计算单元(CUDA CORE),大幅提高运算性能。
在固定邻居算法中,数据结构主要包括各个原子的位置、速度、力等属性,在三维空间中,它们都是以四维向量方式定义的,四个分量x、y、z、w分别表示在三个方向上的方位坐标和空值。线程对数据结构的重映射示意图如图4 所示。float4 数据结构如图4(a)所示,原子属性以四维向量x、y、z、w的形式连续存储在全局内存中,从三个维度分别计算原子间的距离和受力。当计算x方向上两个原子间的距离和受力时,在warp 中,如Thread 0 访问x1,Thread 1访问x2,x1与x2之间相隔y1、z1、w1,两个线程对原子数据的访问和计算会造成计算吞吐量的降低。在图4(b)中,结合Tesla V100 GPU 的硬件特性,修改算法数据结构布局,使原子属性以单位标量x、y、z、w的形式各自分开存储在全局内存中,在计算原子间x方向上的距离和受力时,在warp 中,Thread 0 访问x1,Thread 1 访问x2,类似操作实现了线程对原子数据的连续访问。这种新的数据结构布局方式有助于充分利用硬件特性,提升计算吞吐量和访存带宽性能,提高算法的运行效率。

图4 线程对数据结构的重映射示意图Fig.4 Remapping schematic diagram of data structures by threads
3.2 分支结构优化
GPU 分支结构执行方式不同于CPU 分支结构执行方式。在CPU 中设置分支预测机制,有效缩短执行时间。在GPU 执行中,如果warp 中任何一个线程进入到不同的分支路径,则其余的线程都处于等待状态,直到该线程执行完分支程序。warp 中线程按顺序串行通过多条不同分支路径,当所有分支路径都执行后,warp 中的所有线程才会回到同一条执行路径上。
warp 分支结构如图5 所示。一个线程束warp 包含32 个线程,在warp 内以SIMT 的方式执行[17]。在执行if 分支时,当前18 个线程指向的数据执行时,剩余后14 个线程指向的数据都需要等待,造成线程资源浪费。若程序中的分支路径较多,且warp 中的线程都需要执行时,会延长程序的执行时间,造成性能的严重损失。
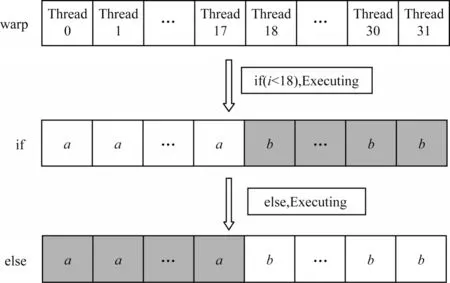
图5 warp 分支结构Fig.5 Structure of warp branch
大多数对于分支结构的优化保留在线程层,线程交换是通过减少同一个warp 中不同分支的方式来提高程序并行性,根据分支控制数据的依赖关系,重新排列数据数组,建立线程与数据的重映射关系,从而提升性能[18-20]。然而,一些特定算法的分支结构无法通过上述方式进行优化,本文引入三目运算符的方法对分支结构进行优化。分支结构转换为三目运算符的示意图如图6 所示。
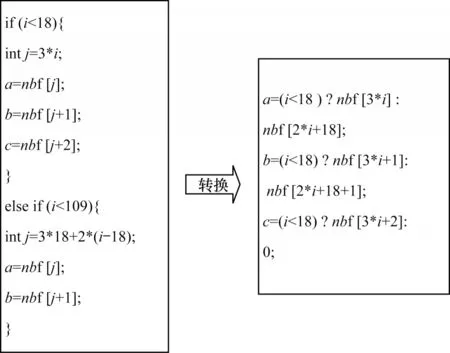
图6 分支结构转换为三目运算符的示意图Fig.6 Schematic diagram of branch structure converted to the ternary operator
固定邻居算法在超胞中的原子受力需要分组赋值的情况下,采用三目运算符代替分支结构。在执行分支结构代码时,if-else 语句会对分支指令进行排队处理,而三目运算符对分支的两种情况同时进行预处理,使三目运算符代码的耗时周期相较于分支结构代码更短,有效减少分支耗时并提升算法的运行效率。
4 计算验证与分析
4.1 计算环境
本文固态共价晶体硅的MD 模拟是在Tesla V100 GPU 单卡上实现并行执行。固定邻居算法的应用环境如表1 所示。采用算法性能最优的超胞尺寸,其包含256 个硅原子,在x、y、z方向上长度分别为2、4、4 个单位晶胞,模拟的初始温度为300 K。
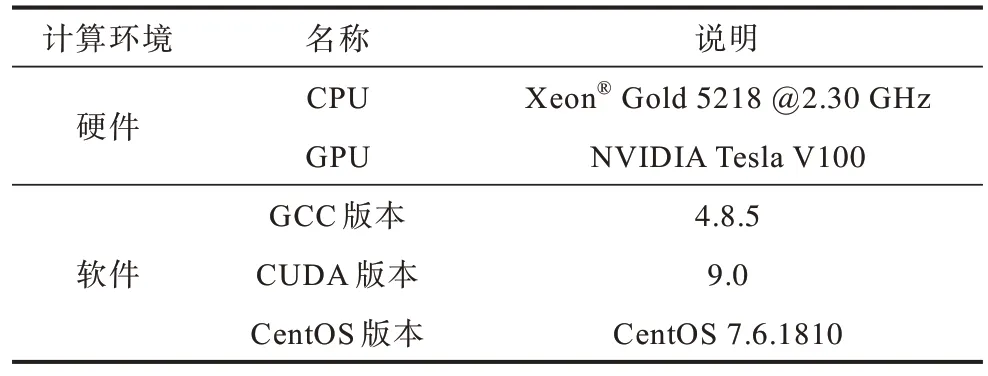
表1 固定邻居算法的计算环境Table 1 Computational environment of fixed neighbor algorithm
4.2 计算测试分析
4.2.1 正确性分析
本文通过算例验证固定邻居算法优化前后的有效性和正确性,总的迭代步数设置为100 万步,原子模拟数目为32 768 个,每间隔1 000 迭代步,输出一次统计结果。GPU 固定邻居算法优化前后每个原子的平均势能随时间变化曲线如图7 所示,SP 表示单精度,DP 表示双精度。

图7 GPU 固定邻居算法优化前后每个原子的平均势能随时间变化曲线Fig.7 Time varying curves of average potential energy of each atom before and after optimizing the fixed neighbor algorithm on GPU
在Tesla V100 GPU 上固定邻居算法优化前后原子的平均势能变化稳定,双精度的原子势能趋向于-4.610 24 eV,单精度的原子势能趋向于-4.610 20 eV,模拟结果与文献[7]一致,证明了固定邻居算法优化前后的计算正确可靠。
4.2.2 性能测试与分析
在超胞尺寸固定不变的情况下,本文考虑到Tesla V100 GPU 单卡硬件资源的限制,分别对不同原子数目进行模拟,实验选择原子数目为32 768、262 144、1 024 000、4 096 000、10 240 000,以模拟1 000 迭代步的运行时间作为评价算法性能的指标,通过对比算法优化前后的耗时来验证优化效果。
将数据结构与分支结构优化方法相结合,固定邻居算法优化前后的性能对比如表2 所示。t1表示双精度固定邻居算法优化前的运行时间,t2表示双精度固定邻居算法优化后的运行时间,t3表示单精度固定邻居算法优化前的运行时间,t4表示单精度固定邻居算法优化后的运行时间。当原子数目为4 096 000时,优化后双精度固定邻居算法的性能最优,与固定邻居算法优化前的运行时间相比,双精度固定邻居算法与单精度固定邻居算法的加速效果分别提高了27.7%与20.7%。
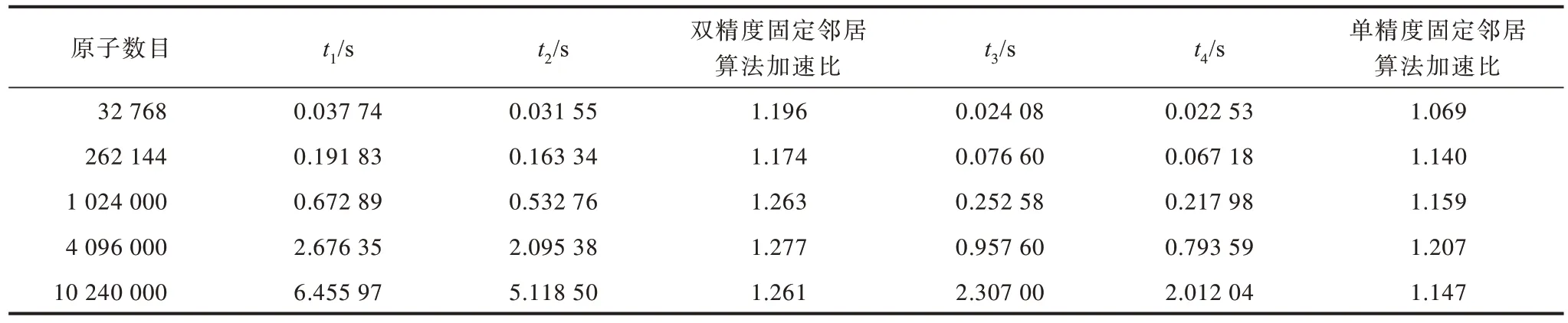
表2 固定邻居算法优化前后性能对比Table 2 Performance comparison of fixed neighbor algorithms before and after optimization
固定邻居算法优化后的加速比如图8 所示。当原子数目为32 768 时,优化后双精度固定邻居算法加速比是优化后单精度固定邻居算法加速比的1 倍多。其原因为当原子规模较小时,优化后双精度固定邻居算法计算1 000 迭代步的运行时间较短。此时,单精度固定邻居算法计算力的核函数耗时占GPU 端总耗时的比例较小,双精度固定邻居算法计算力的核函数耗时占GPU 端总耗时比例较大,因此,优化后双精度固定邻居算法的加速比较高。固定邻居算法从原子数目为262 144 开始,优化后单精度和双精度固定邻居算法的加速比随着原子数目的增大而增大,并在硬件资源限制的条件下达到一个最高值,然后,加速比开始下降。
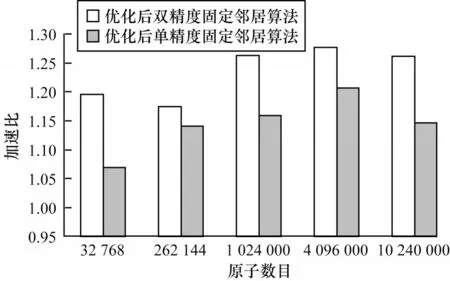
图8 固定邻居算法优化后的加速比Fig.8 Acceleration ratio of fixed neighbor algorithm after optimization
在Tesla V100 GPU 处理器资源可承受范围内,固定邻居算法的优化效果依赖于计算的浮点精度和计算规模。在GPU 计算资源和存储资源相同的条件下,原子数目多的体系优化效果更好。其原因为当原子数目较小时,浮点计算不能充分利用Tesla V100 GPU 处理器并行资源,计算性能不能达到最优,当原子数目较大时,Tesla V100 GPU 处理器才能充分发挥计算优势,加速效果明显。
4.3 与LAMMPS和HOOMD-blue软件的性能对比
LAMMPS 是一种支持多种原子间势函数并被广泛应用的开源MD 软件,利用空间分解技术将三维模拟区域划分为更小的三维子域,利用MPI 并行化提高计算规模和性能。KOKKOS 加速包是基于LAMMPS 的C++加速库,可以有效地在GPU 硬件上运行[21]。目前,KOKKOS 加速包没有精度选项,所有编译和计算都必须以双精度执行[22-23]。HOOMDblue 是专门支持GPU 加速的分子动力学模拟软件,能够在多种力场下执行MD 模拟计算[24-25]。本文使用LAMMPS 软件KOKKOS 加速包和支持GPU 加速的HOOMD-blue 软件在Tesla V100 GPU 上对Tersoff多体势进行单双精度计算,测试环境与4.1 节相同,选择模拟1 000 迭代步的运行时间作为性能对比的基准。
LAMMPS 和HOOMD-blue 与优化后的固定邻居算法性能对比如表3 所示,表中T1表示双精度固定邻居算法的执行时间,T2表示单精度固定邻居算法的执行时间,T3表示LAMMPS 双精度固态晶体硅分子动力学模拟的执行时间,T4表示HOOMD-blue 双精度固态晶体硅分子动力学模拟的执行时间,T5表示HOOMDblue 单精度固态晶体硅分子动力学模拟的执行时间,加速比1 表示LAMMPS 双精度固态晶体硅分子动力学模拟与双精度固定邻居算法(LAMMPS 双精度/双精度算法)的执行时间比值,加速比2 表示HOOMD-blue双精度固态晶体硅分子动力学模拟与双精度固定邻居算法(HOOMD-blue 双精度/双精度算法)的执行时间比值,加速比3 表示HOOMD-blue 单精度固态晶体硅分子动力学模拟与单精度固定邻居算法(HOOMD-blue单精度/单精度算法)的执行时间比值。
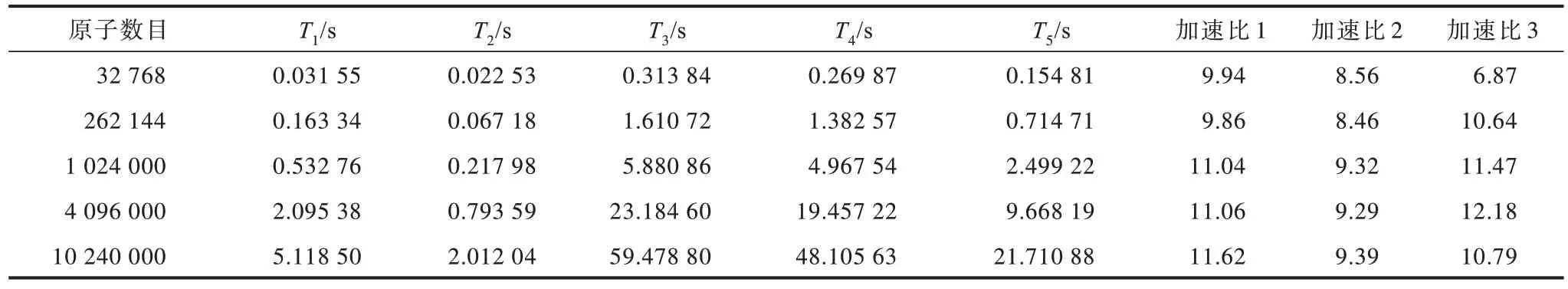
表3 LAMMPS 和HOOMD-blue 与优化后的固定邻居算法性能对比Table 3 Performance comparison of LAMMPS and HOOMD-blue with the optimized fixed neighbor algorithm
相比两个MD 开源模拟软件,固定邻居算法具有较优的加速效果。当原子数目为10 240 000 时,双精度固定邻居算法加速效果最优,LAMMPS 和 HOOMD-blue 软件与双精度固定邻居算法的执行时间的比值分别为 11.62 和 9.39。当原子数目为 4 096 000 时,单精度固定邻居算法的加速效果较优,HOOMD-blue 软件与单精度固定邻居算法执行时间的比值为12.18。优化后的固定邻居算法相对LAMMPS 和HOOMD-blue 的加速比如图9 所示。随着原子数目的增加,双精度固定邻居算法与两个 MD 开源模拟软件相比,其加速比呈现上升趋势。HOOMD-blue 单精度/单精度算法加速比呈现先升高后略微下降的趋势。

图9 优化后的固定邻居算法相对LAMMPS 和HOOMD-blue 的加速比Fig.9 Acceleration ratio of the optimized fixed neighbor algorithm to LAMMPS and HOOMD-blue
5 结束语
针对固态共价晶体硅的分子动力学模拟,本文结合GPU 的硬件架构特性,提出分子动力学模拟固定邻居算法的优化设计,采用数据结构和分支结构优化方法,减少多线程访存和分支耗时,提升晶体硅模拟的计算效率。与主要的GPU 加速MD 模拟软件LAMMPS 和HOOMD-blue 相比,本文固定邻居算法具有较优的加速性能。下一步将对多GPU 并行的分子动力模拟算法进行研究,有效提升固相晶体硅体系的计算规模,实现数十亿原子以上的模拟。
