小麦苗期根系性状的全基因组关联分析与优异位点挖掘
2023-03-14王脉董清峰高珅奥刘德政卢山乔朋放陈亮胡银岗
王脉,董清峰,高珅奥,刘德政,卢山,乔朋放,陈亮,胡银岗,2
小麦苗期根系性状的全基因组关联分析与优异位点挖掘
王脉1,董清峰1,高珅奥1,刘德政1,卢山1,乔朋放1,陈亮1,胡银岗1,2
1西北农林科技大学农学院/旱区作物逆境生物学国家重点实验室,陕西杨凌 712100;2中国旱区节水农业研究院,陕西杨凌 712100
【目的】植物根系对水分及营养的获取、作物的生长发育和产量的形成至关重要。挖掘小麦苗期根系性状显著关联的SNP位点,预测相关候选基因,为解析小麦根系建成遗传机制及选育具有优良根系构型的小麦品种奠定基础。【方法】以189份小麦品种组成的自然群体为供试材料,调查2种培养条件(霍格兰营养液和去离子水)下培育21 d的苗期根系总长度(TRL)、根系总表面积(TRA)、根系总体积(TRV)、根系平均直径(ARD)及根系干重(RDW)等5个根系性状,试验进行2次重复,同时结合小麦660K SNP芯片的分型结果进行全基因组关联分析(genome-wide association study,GWAS)。此外,通过序列比对、结构域分析和注释信息预测候选基因,并采用竞争性等位基因(kompetitive allele specific PCR,KASP)技术开发根系性状的分子标记。【结果】霍格兰营养液培养条件下的根系性状变异范围较大,根系整体粗短;而去离子水条件下的根系细长、侧根较多。选用贝叶斯信息与连锁不平衡迭代嵌套式模型(BLINK)、压缩式混合线性模型(CMLM)、固定随机循环概率模型(FarmCPU)以及混合线性模型(MLM)4个模型,结合2种培养条件下的根系性状进行全基因关联分析,共检测到95个与小麦苗期根系性状显著关联的QTL位点(<10-3),其中,有18个QTL在2个条件下同时被检测到,分布在7A、1B、2B、3B、7B、1D、2D及3D染色体,可解释8.68%—14.07%的表型变异。筛选获得的显著性位点中,有4个与前人的研究相近或一致,其余为新发现QTL位点。对共定位的SNP进行单倍型分析,有10个SNP能够将供试材料分为2种单倍型,且单倍型间的根系性状具有显著差异,同时,基于这些SNP开发KASP标记,筛选到与根系总体积及根系干重相关的2个KASP标记(和)。进一步挖掘共定位SNP位点上下游区间内的基因,筛选到12个可能与根系发育相关的候选基因,其中,编码酰基载体蛋白合成酶,参与根系脂肪酸的合成;编码突触融合蛋白,对植物重力向性具有重要作用;编码醛脱氢酶,参与脱落酸的合成,从而调控作物根系发育。【结论】小麦根系性状在不同基因型间差异显著,在2个条件下同时检测到18个显著QTL位点,开发了2个根系分子标记(和),并筛选出12个与根系性状相关的候选基因。
小麦;根系性状;全基因组关联分析;共定位SNP位点;KASP标记;候选基因
0 引言
【研究意义】小麦(L.)是世界三大主粮作物之一,全球小麦种植面积2.17亿hm2,年产量约7.65亿t[1],为人类提供近20%的热量。近年来,随着全球人口的急剧增长,人类对小麦的需求也随之增加。据联合国农业组织预测:到2050年,每年至少需要生产8.4亿t小麦[2]。因此,提高小麦单产对保障全球粮食生产具有重要作用。根系作为植物三大器官之一,是植物吸收矿物质养分及水分的第一部位,对植物生长起着固定和支撑作用[3]。根系建成(root system architecture,RSA)包括根系长度、根系表面积、根系体积、根系直径、根尖数以及根系倾角等根系性状[4],根系构型的好坏是决定作物生长发育和提高小麦产量及抗逆性的必要因素。解析作物根系相关性状的遗传机制,优化作物根系构型对小麦育种具有重要意义[4-6]。【前人研究进展】近年来,国内外研究人员在根系研究方面做了大量工作,并已取得阶段性成果。研究表明[7],优良的根系构型可响应各种生物与非生物胁迫,通过信号转导途径影响地上部的生长,与千粒重、粒长、粒宽等产量性状显著正相关。Xie等[8]研究发现根长较短的小麦品种穗数、穗粒数和植株生物量均较低,而根长较长的品系生物量和籽粒产量较高。Man等[9]研究指出小麦根系干重与籽粒产量和水分利用效率呈正相关关系。此外,随着基因组学和分子生物学研究的不断深入,分子育种的优势逐渐凸显。目前,利用小麦不同遗传群体已经定位到了许多与根系性状相关的位点。如,Ma等[10]利用388份小麦品种在室内和室外盆栽2种环境下对根系性状进行全基因组关联分析(genome-wide association study,GWAS),在2A、2B、3A、3B、3D、4A、5B、5D、6B染色体共检测到36个和根系建成显著关联的QTL位点;陈贵菊等[11]对160份来自于河南和山东等地小麦品种的总根长、总根表面积、总根体积、平均根直径和根尖数进行全基因组关联分析,检测到23个关联位点,可解释7.2%—12.8%的表型变异;Huang等[12]通过对周8425B×中国春构建的重组自交系群体进行根毛长度的QTL分析,检测到4个主效QTL位点(LOD>2.5),解释3.32%—6.52%的表型方差。Khodaee等[13]将170份春小麦材料种植于土壤的PVC管里,对水分胁迫和正常供水条件下的根系长度及根系体积等性状进行全基因组关联分析,鉴定出186个显著性SNP位点,其中一些SNP证实了先前报道的根长位点。同时,由于SNP在基因组分布广泛且稳定性高,基于SNP检测的竞争性等位基因(kompetitive allele specific PCR,KASP)技术已普遍用于小麦抗病性和抗非生物胁迫相关基因的检测和辅助选择[14-16]。Qureshi等[17]借助20对KASP标记证实了小麦抗条锈病和为同一个基因。Fang等[18]开发了2对小麦抗叶锈基因的KASP标记:和,以加速小麦育种中对的辅助选择。Rehman等[19]开发了16个与小麦抗旱相关基因座等位基因的KASP标记,分析了47份小麦材料的等位变异与籽粒产量的相关性。王志伟等[20]利用3对抗旱及抗穗发芽基因的KASP标记,对云南育成的小麦品种(系)进行基因型检测,表明其能够有效区分抗穗发芽和易穗发芽的品种及抗旱和非抗旱品种。【本研究切入点】鉴于小麦根系的难获取性及数量性状遗传调控的复杂性,目前得到的小麦根系构型相关位点的数量并不多,因此,亟需扩大基因组筛选的广度和深度,增加分子标记的数目及种类,挖掘更多主效QTL,从而对调控根系发育的基因进行精细定位。【拟解决的关键问题】本研究以189份小麦品种(系)组成的自然群体为材料,在霍格兰营养液和去离子水2种培养条件下,测定苗期根系的总根长、根系总表面积、根系总体积、根系平均直径及根系干重(与根系构型相关的性状);进一步结合高通量660K SNP芯片的基因型分析结果,对根系性状进行全基因组关联分析,以期发掘调控小麦根系性状的QTL位点,开发根系分子标记,并预测与根系构型相关的候选基因,为小麦根系改良及抗逆育种奠定基础。
1 材料与方法
1.1 试验材料
以实验室通过多年试验,并结合35K芯片进行基因型分析的565份小麦种质中筛选出189份小麦品种(系)组成的自然群体为材料,这些品种遗传多样性丰富,来源广泛,主要来自河南(73份)、陕西(51份)、山东(21份)、甘肃(16份),以及河北、江苏、宁夏、山西、北京、安徽等地,还包括部分从澳大利亚、美国及墨西哥引进的种质(电子附表1)。
1.2 试验设计
供试材料于2021年7—8月及2022年1—2月在西北农林科技大学旱区逆境生物学国家重点实验室光照培养间进行水培试验,分别采用霍格兰营养液(hogland,HL)和去离子水(pure water,PW)进行培养,试验均重复2次。选用大小均匀一致的小麦种子,采用75%的酒精消毒1 min,用蒸馏水冲洗,摆放在培养皿中进行发芽。幼苗长至1叶1心期时(约7 d),每个品种选择长势一致的4株幼苗,移栽并定植于泡沫板上,每孔种植1粒,放置于根箱中(50 cm×40 cm×30 cm),20℃恒温培养,光照16 h/黑暗8 h,湿度为70%。每天通过氧气泵间断通气10 h,培养21 d。期间每隔3 d更换1次营养液和去离子水,营养液用稀NaOH和0.1 mmol·L-1HCl调节pH至6.5。
1.3 测定指标及测定方法
植株水培21 d后,使用EPSON Perfection V700 Photo扫描仪扫描根系存成图片,然后通过万深LA-S根系分析仪系统(www.wseen.com)处理并分析扫描的图片,得到根系总长度(total root length,TRL,cm)、根系总表面积(total root area,TRA,cm2)、根系总体积(total root volume,TRV,cm3)、根系平均直径(average root diameter,ARD,mm)等参数。扫描后的根系105℃杀青30 min后,80℃烘干至恒重,用万分之一天平测定根系干重(root drought weight,RDW,g)。
1.4 表型数据的统计分析
采用Excel 2019和IBM SPSS Statistics 25(https://www.ibm.com/cn-zh/spss)对根系总长度、根系总表面积、根系总体积、根系平均直径及根系干重等5个性状进行统计分析,分别计算2个培养条件下各根系性状的平均值、标准差、变异系数以及广义遗传力。运用R语言绘制各根系性状的频率分布图,利用Origin 2021(https://www.originlab.com/origin)软件对各根系性状进行相关性分析。
1.5 群体结构与连锁不平衡分析
采用改良的CTAB法提取小麦幼嫩叶片的总基因组DNA,质检合格后,由北京博奥生物技术有限公司利用小麦660K SNP芯片对189个小麦品种进行基因分型,并采用TASSEL 5.0软件[21]对原始数据进行质量控制和过滤,剔除缺失率和杂合度>20%,次等位基因频率<5%[22]的SNP位点。基于过滤后的高质量SNP标记,采用Power Marker V3.25软件[23]分析供试材料的遗传多样性和多态信息含量(polymorphic information content,),=1-Σ(P)2(P表示位点的第个等位变异出现的频率)。进一步使用Admixture软件[24]进行群体结构分析:令K=2—15,对交叉验证错误率CV值(cross-validation error)进行5次训练,最终将5个模型的预测值做平均,CVmin对应的K值即为最佳亚群数目。采用Origin 2021对189份小麦材料进行主成分分析(principal component analysis,PCA),并绘制亚群的3D图。以位点间的相关系数平方(2)为参数衡量多态性位点两两之间的连锁不平衡度(linkage disequilibrium,LD),使用PopLDdecay软件[25]进行ABD基因组的连锁不平衡分析,2的参数设置为:-MaxDist30000[25-26]。将30 000 kb LD区间划分为10 kb的区间,以第95百分位的2值作为阈值估测LD衰减距离。
1.6 全基因组关联分析与候选基因挖掘
通过对小麦660K SNP芯片的基因分型结果进行质控和过滤,最终获得296 111个高质量SNP标记,用于后续的全基因组关联分析。通过基于R语言的Gapit包(http;//www.zzlab.net/GAPIT),选用BLINK(bayesian-information and linkage-disequilibrium iteratively nested keyway)、CMLM(compressed mixed linear model)、FarmCPU(fixed and random model circulating probability unification)和MLM(mixed linear model)4个模型进行全基因组关联分析,以=1.0×10-3为阈值,判定与目标性状显著关联的SNP位点。基于共定位的SNP位点,将供试品种分为2种单倍型,采用SPSS的ANOVA检测比较2种单倍型间目标根系性状的差异显著性。运用Origin 2021绘制单倍型分析的箱线图。采用R包的CMPlot绘制QQ图和曼哈顿图,并在共定位SNP位点LD衰减范围内挖掘小麦根系性状的候选基因。在NCBI数据库(https://blast.ncbi.nlm.nih.gov)、InterPro(https://www.ebi.ac.uk/interpro)、WheatOmics(http://wheatomics.sdau.edu.cn)和Ensembl plants(http://plants.ensembl.org/index.html)等网站进行候选基因的序列比对、保守结构域分析和功能注释。利用expVIP(http://www.wheat-expression.com)获取小麦候选基因在不同组织的表达量,并运用TBtools软件[27]绘制基因表达量热图。
1.7 显著性SNP分子标记开发及检测
根据660K SNP芯片的遗传图谱信息,基于与根系性状显著关联的SNP位点开发KASP标记。基于在线网站Poly Marker(http://www.polymarker.info)设计KASP引物(表1),在2条正向引物序列的5′端分别加上FAM和HEX荧光接头序列,其中,FAM接头序列为:5′-GAAGGTGACCAAGTTCATGCT-3′,HEX(VIC)为:5′-GAAGGTCGGAGTCAACGGAT T-3′。引物由北京擎科生物科技有限公司合成。
利用根系性状极端的小麦材料进行引物的初步筛选,筛选出具有多态性且分群明显的KASP标记(和X),进一步在189份小麦品种组成的自然群体中进行基因型分析。KASP分型系统操作参考北京嘉程生物科技有限公司的AQP基因分型说明:384孔板反应体系包含Higeno 2×Probe Mix B 2.5 μL、引物Mix 0.07 μL(2条正向引物12 mmol·L-1,反向引物30 mmol·L-1)、DNA模板1 μL,ddH2O补至5 μL。PCR反应采用touchdown程序:95℃ 10 min;95℃ 20 s,61—55℃ 40 s,10个循环(每个循环降0.6℃);95℃20 s,55℃ 40 s,32个循环,共计42个循环。反应完成后读取荧光数据,通过Klustercaller SNP分型软件检测分型结果。
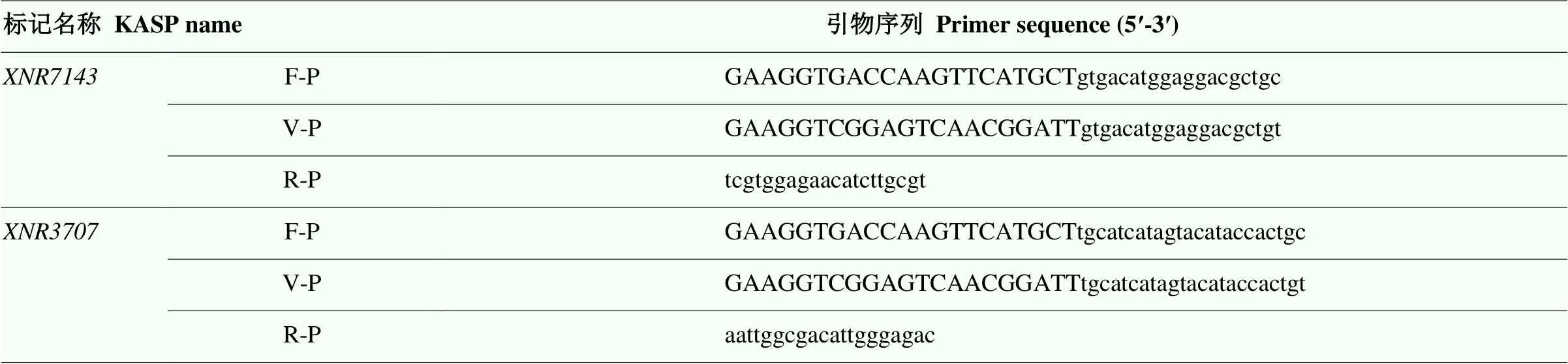
表1 多态性KASP标记的引物序列
F-P:连接FAM荧光序列的正向引物;V-P:连接HEX(VIC)荧光序列的正向引物;R-P:反向引物
F-P: Forward primer with FAM fluorescence sequence; V-P: Forward primer with HEX(VIC) fluorescence sequence; R-P: Reverse primer
2 结果
2.1 不同环境下小麦苗期根系性状的表型分析
通过对霍格兰营养液(HL)和去离子水(PW)2个培养条件下供试品种的根系总长度、根系总表面积、根系总体积、根系平均直径以及根系干重等性状进行统计分析(电子附表2),发现2个培养条件下各根系性状的频率呈正态分布(图1),表明各根系性状是数量性状,受多基因控制,其表型数据可用于后续的全基因组关联分析。2种培养条件下的各根系性状均表现出广泛变异,变异范围为15.43%—66.38%(表2),其中,在HL培养下的各根系性状变异幅度较大,PW培养下的变幅相对较小。2个培养条件下的根系性状差异明显,HL培养下的根系总长度、根系总表面积及根系干重均显著低于PW,而根系平均直径及根系总体积高于PW。方差分析表明,各根系性状在小麦品种间、培养条件间以及品种与培养条件互作下均呈极显著差异。各根系性状的广义遗传力为60.41%— 65.30%,其中,根系干重的广义遗传力最大(表3),表明根系性状的遗传稳定性较强。
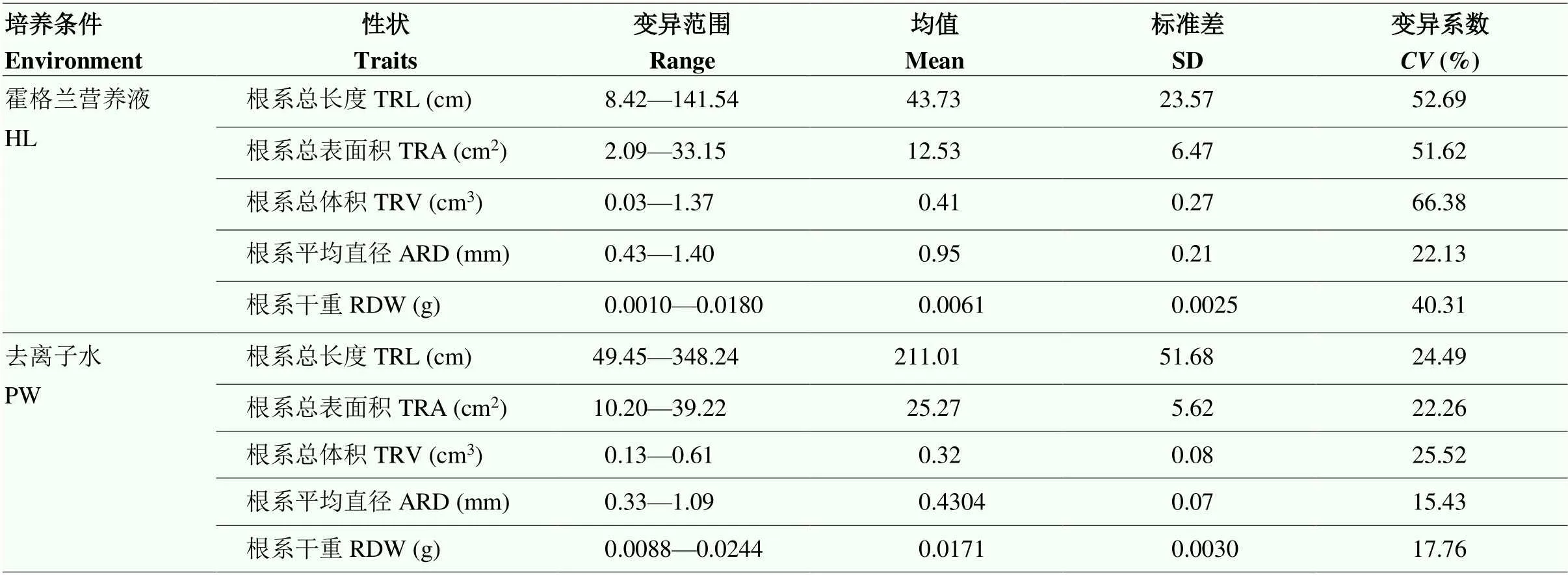
表2 不同处理下小麦种质各根系性状的描述统计分析
HL:霍格兰营养液;PW:去离子水;TRL:根系总长度;TRA:根系总表面积;TRV:根系总体积;ARD:根系平均直径;RDW:根系干重。下同
HL: hogland; PW: pure water; TRL: total root length; TRA: total root area; TRV: total root volume; ARD: average root diameter; RDW: root dry weight. The same as below
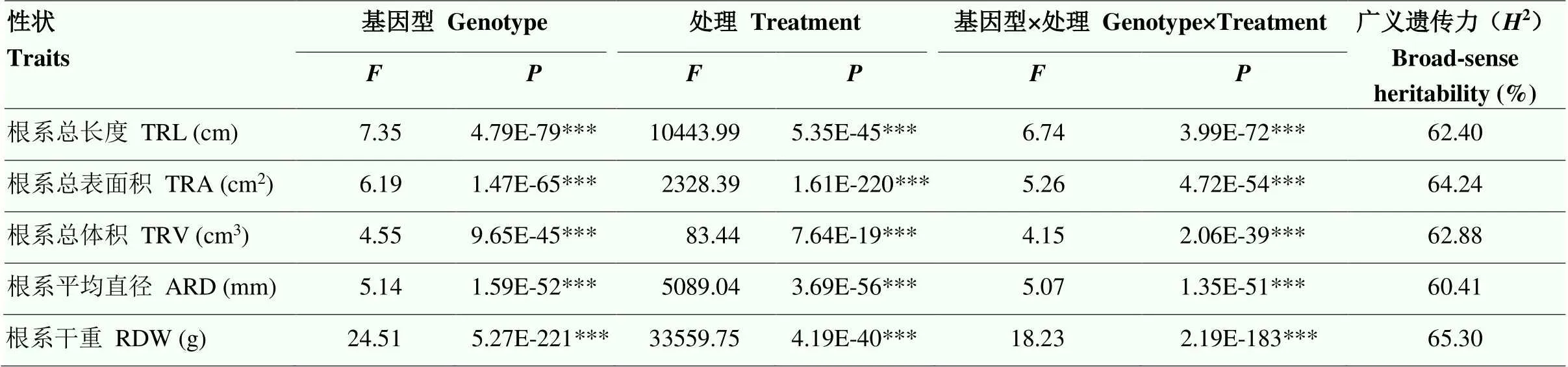
表3 小麦种质各根系性状的方差分析及广义遗传力
***表示在<0.001水平差异显著 *** represents significance of difference at<0.001
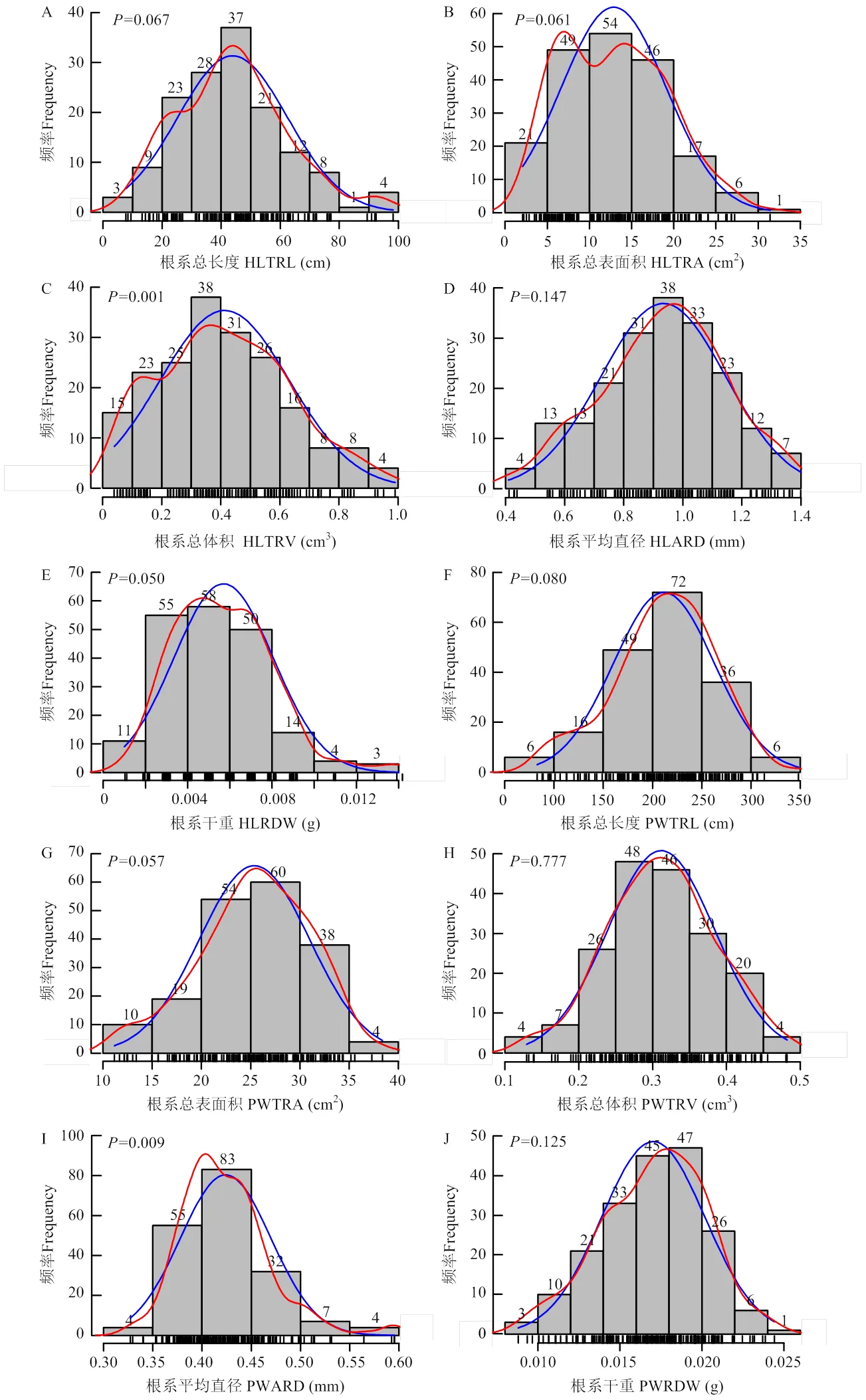
A—E:霍格兰营养液培养;F—J:去离子水培养 A-E: Hogland culture; F-J: Pure water culture
相关分析表明(图2),在HL和PW 2种培养条件下的根系总长度、根系总表面积、根系总体积和根系干重等根系性状之间呈极显著正相关。HL条件下的根系总体积、根系总长度和根系总表面积的相关系数达到0.92和0.81,而根系总长度和根系平均直径呈负相关;PW条件下的根系总长度、根系总体积与根系总表面积的相关系数为0.96和0.90,而根系总长度与根系平均直径呈极显著负相关,根系干重及根系平均直径呈显著负相关。总之,2种培养条件下的各根系性状间的相关性基本一致。
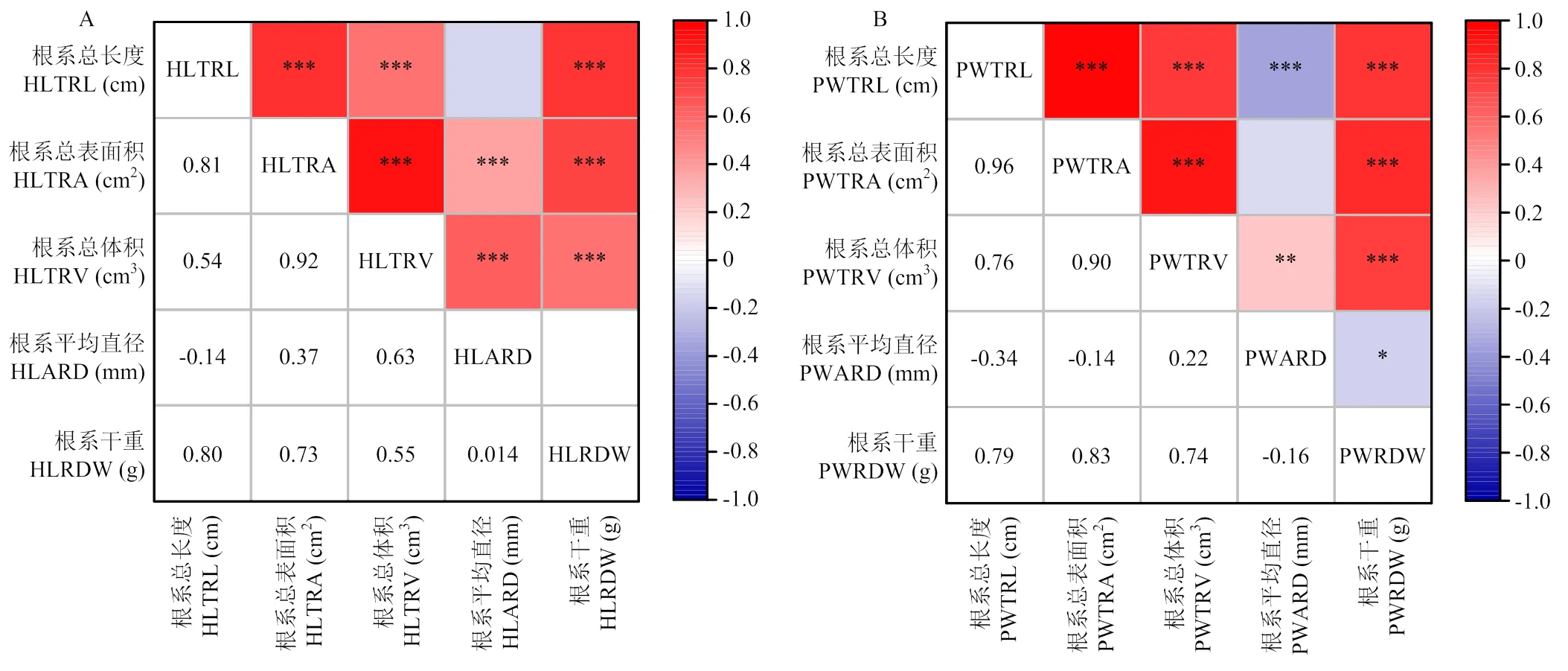
A:霍格兰营养液培养;B:去离子水培养。*、**和***分别表示在P<0.05、P<0.01以及P<0.001水平差异显著。下同
2.2 SNP分布及多态性分析
利用CMplot对660K SNP标记在染色体的位置进行绘制,发现SNP标记基本覆盖所有染色体区段,然而这些标记在各染色体的分布并不均匀,总体表现出“两边多中间少”的趋势(图3)。通过对获得的SNP数据进行高质量质控,最终筛选出296 111个具有遗传多样性的SNP标记,其在A、B、D基因组的数目分别为115 694、140 634和39 783个,分别占总基因组的39.1%、47.5%和13.4%(表4);其中,3B染色体的SNP最多(36 612个),而4D染色体的SNP最少(2 408个)。A、B基因组的遗传多样性分别为0.3575和0.3660,均大于D基因组。全基因组的多态信息量均值为0.2858,各亚基因组的多态信息量表现为B基因组(0.2926)>A基因组(0.2860)>D基因组(0.2789),说明B基因组的等位基因频率更平等。
2.3 群体结构与连锁不平衡分析
基于Admixture软件对群体结构进行分析的结果表明,K=8时,CV error达到最小值,因此,可将189份小麦材料分为8个类群,将K=8生成的Q矩阵作为协变量用于后续的全基因组关联分析(图4-A和图4-B)。进一步对189份小麦材料进行主成分分析,其中,前3个主成分分别可解释表型变异的17.79%、16.09%以及14.87%(图4-C)。该189份小麦品种(系)大多来自中国西北、华中及华东地区,其中,亚群Ⅰ有25个(13%)品种,主要包含陕西和甘肃品种;亚群Ⅱ和亚群Ⅲ分别有25个(13%)和10个(5%)品种,均主要来自河南;亚群Ⅳ有34个(18%)品种,主要包含陕西品种及少量河南品种;亚群Ⅴ有37个(20%)品种,主要为山东和河南品种,还有少量江苏及河北品种;亚群Ⅵ有11个(6%)品种,均来自河南;亚群Ⅶ有37个(20%)品种,主要来自陕西,还有一些品种来自甘肃、河北及河南;亚群Ⅷ有10个(5%)品种,主要来自河南(电子附表1和电子附图1)。通过PopLDdecay对小麦A、B和D亚基因组及全基因组的SNP位点进行连锁不平衡分析表明,随着物理距离的增加,LD明显衰减,但整个基因组和A、B、D亚基因组的衰减存在一定差异(图4-D)。2衰减到0.3时,整个基因组的LD平均衰减距离约为1.5 Mb,而A和B基因组的LD平均衰减距离约为1和3 Mb,D基因组的衰减距离约为0.7 Mb,与小麦进化史上D基因组的快速衰减是一致的[28]。
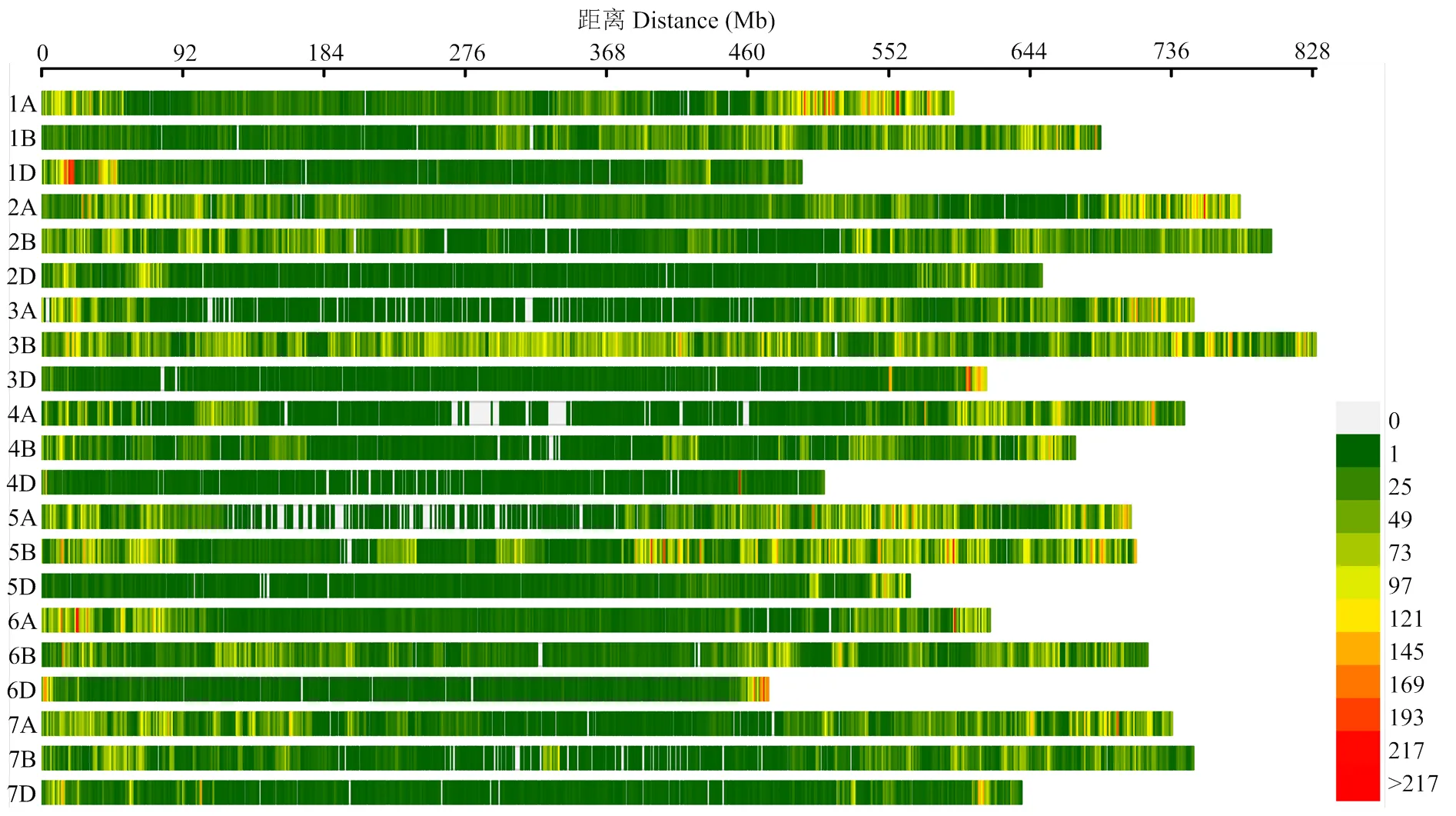
图3 SNP标记在小麦各染色体的分布
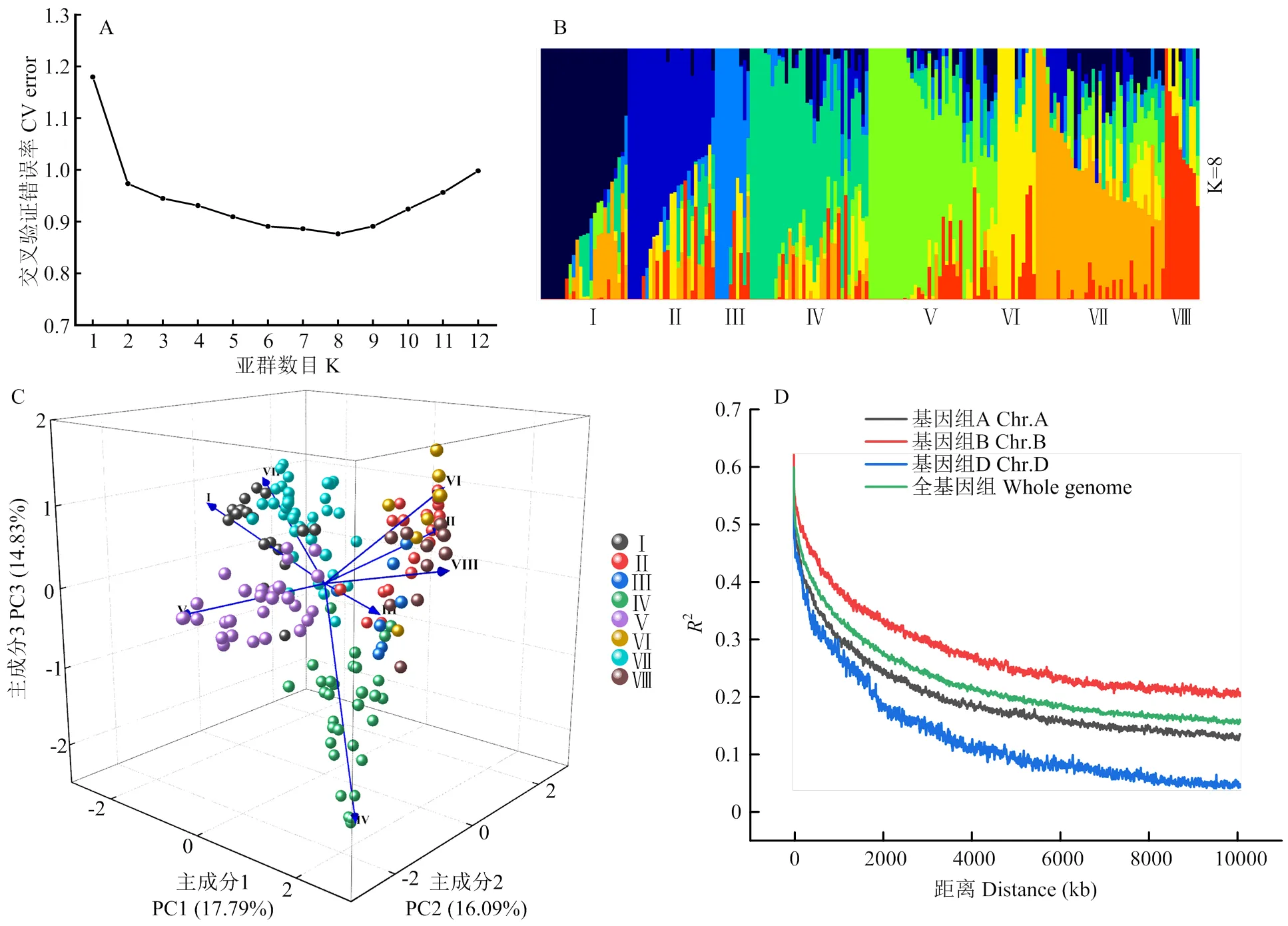
A:亚群的CV error值;B:群体结构示意图;C:189份小麦品种(系)的主成分分析;D:各亚基因组的连锁不平衡衰减距离
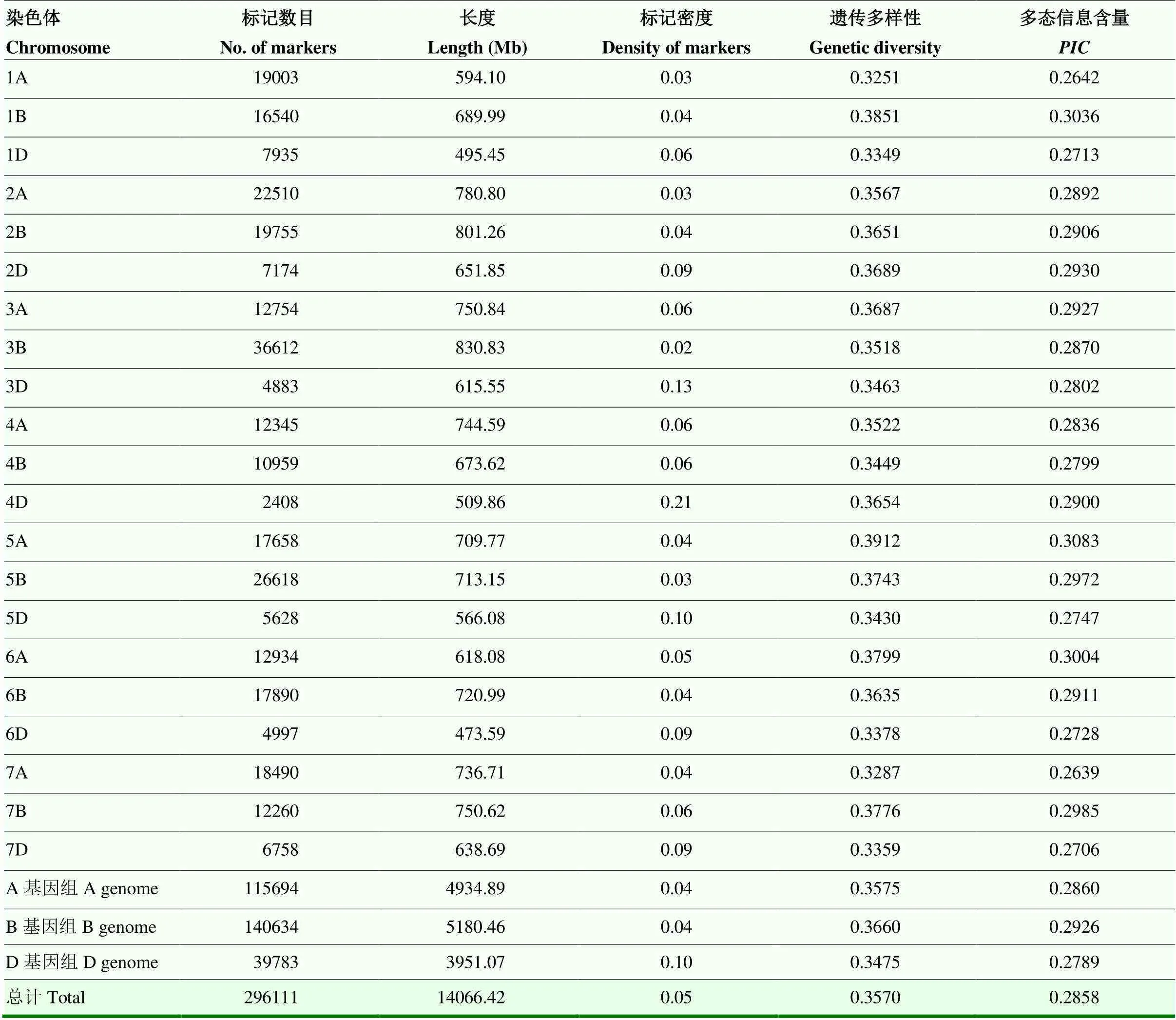
表4 小麦各染色体SNP标记的数目及多态性
2.4 小麦苗期根系性状的全基因组关联分析
利用BLINK、CMLM、FarmCPU以及MLM模型分别对189份小麦群体的根系总长度、根系总表面积、根系总体积、根系平均直径以及根系干重5个根系性状进行全基因组关联分析,在4个模型共有且阈值<10-3的条件下,共检测到2个培养条件与根系性状显著关联的SNP位点95个(图5和电子附表3)。
为了确保关联的可靠性,筛选2个培养条件下同时检测到的18个与小麦根系性状显著关联且稳定遗传的SNP位点进行后续分析(表5),其中,与根系总长度关联的位点5个,与根系干重关联的位点6个,与根系总表面积关联的位点4个,与根系总体积关联的7个,分布在7A、1B、2B、3B、7B、1D、2D和3D染色体,解释了表型变异的8.68%—14.07%,而未检测到在2个培养条件下均与根系直径关联的位点。此外,有些显著性SNP位点同时关联到多个根系性状,如AX-108888527同时关联到根系总长度及根系总表面积,AX-109029863同时关联到根系总长度及根系干重,AX-108792070和AX-110097055关联到根系总表面积及总体积。
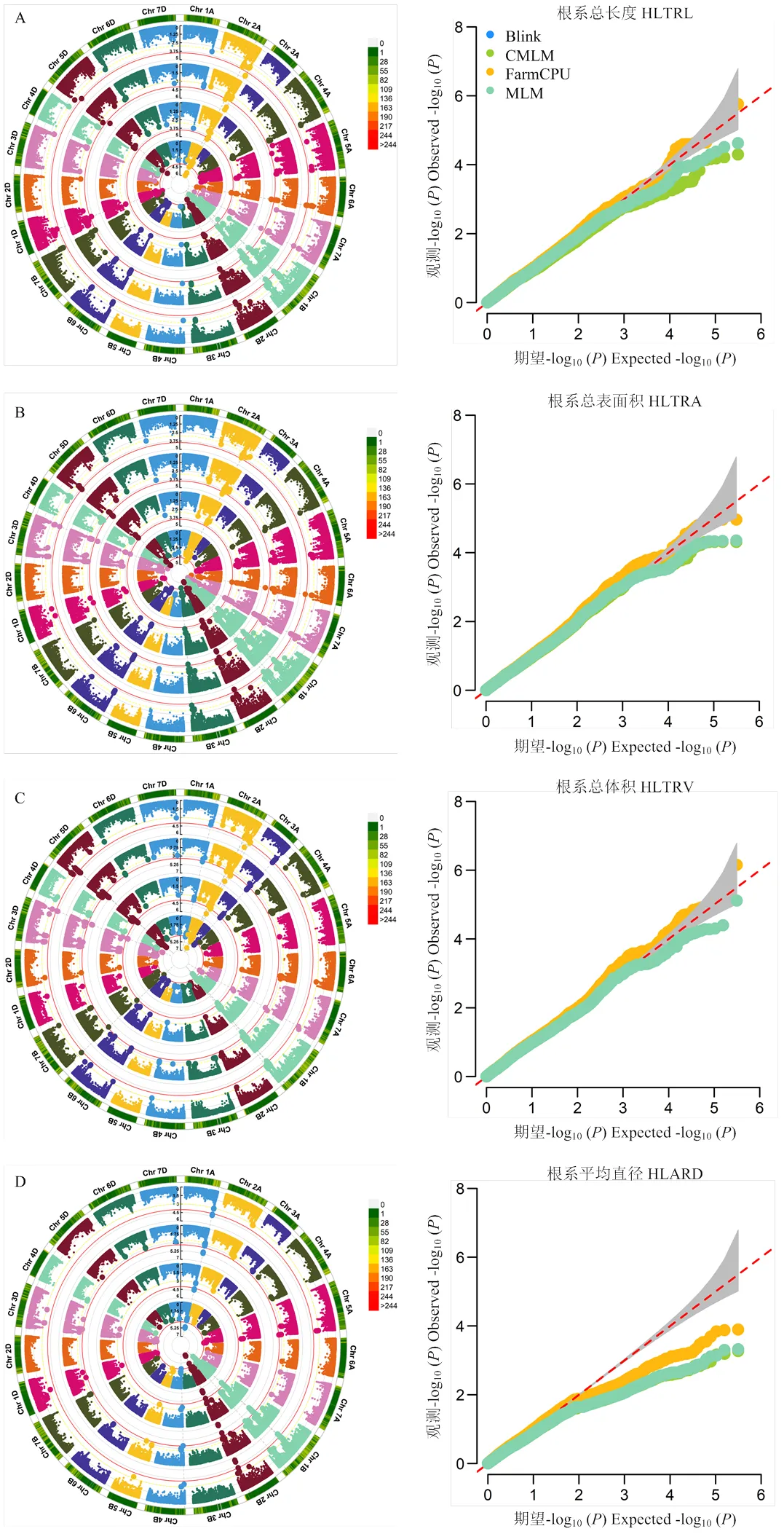
A—E:霍格兰营养液培养;F—J:去离子水培养;曼哈顿图:从里环到外环分别为各根系性状的BLINK、CMLM、FarmCPU以及MLM模型
A-E: Hogland culture; F-J: Pure water culture; Manhattan plots: The BLINK, CMLM, FarmCPU and MLM models of root traits from the inner to outer were respectively showed
图5 不同培养条件下各根系性状的环形曼哈顿图及QQ图
Fig.5 Circular Manhattan plots and QQ plots of root traits in different culture conditions
2.5 小麦苗期根系性状关联位点的单倍型分析
对2个环境中同时检测到的显著性SNP位点进行单倍型分析,其中,5个SNP位点(AX-108924279、AX-110371944、AX-110455550、AX-110567747和AX-109933707)共定位于2B染色体57.144—57.960 Mb的连锁群,与根系干重显著相关(图6-A),其分别处于的外显子和内含子、的下游以及的内含子区域。利用其将189份小麦品种分为2个单倍型(Hap1和Hap2;图6-B),其中Hap1出现的频率为56.8%,Hap2的频率为43.2%。在HL和PW培养条件下,2个单倍型之间的根系干重存在显著和极显著差异(图6-C),Hap2显著提高了小麦苗期的根系干重,是优异单倍型。
此外,除连锁群内的SNP外,进一步对7个单个SNP将供试品种分为2种单倍型进行分析(图7)。其中,2D染色体的AX-108888527位于下游397 bp,与根系总长度及根系总表面积显著关联。在HL培养条件下,2种单倍型的根系总长度及根系总表面积均存在极显著差异;PW培养条件下,2种单倍型的根系总长度存在显著差异,而根系总表面积间差异不显著(图7-A—B)。1B染色体的AX-109555941位于的3端非翻译区,与根系总长度显著关联,2种单倍型间的根系总长度在HL和PW培养条件下存在极显著和显著差异(图7-C)。1B染色体的AX-110491393位于上游616 bp,与根系总体积显著关联,在HL和PW培养条件下,Hap2的根系总体积极显著和显著高于Hap1(图7-D)。1D染色体的AX-94823257位于的外显子区域,与根系总体积显著关联,在HL和PW培养条件下,Hap2的根系总体积分别极显著和显著高于Hap1的根系总体积(图7-E)。2B染色体的AX-111514405位于和之间,与根系总长度显著关联,2种培养条件下Hap1的根系总长度均显著高于Hap2(图7-F)。2B染色体的AX-108889829及7B染色体的AX-110122975分别与根系总长度和根系总体积显著关联。在PW培养条件下,2种单倍型间的根系总长度和根系总体积差异极显著,而在HL培养条件下差异不显著(图7-G—H)。
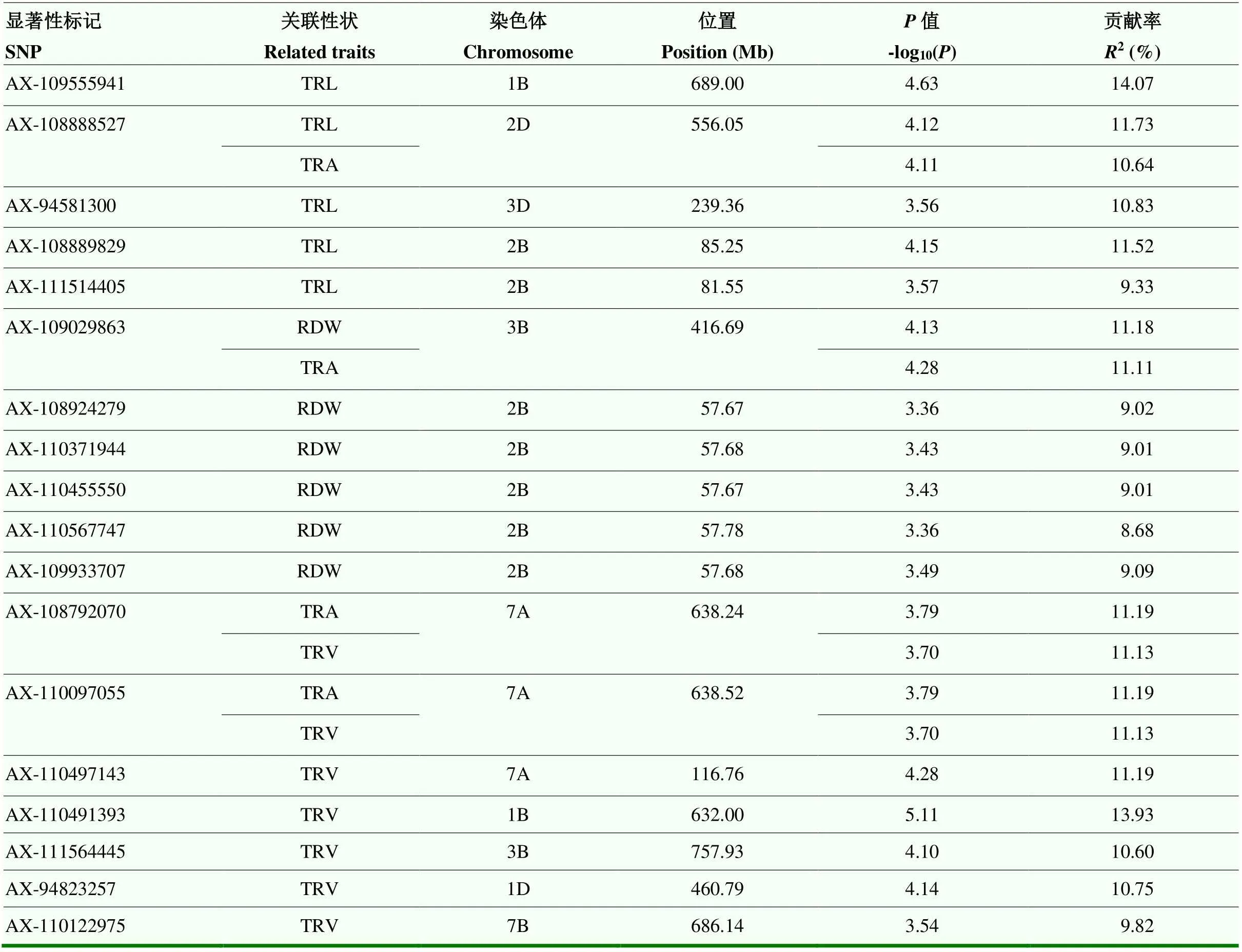
表5 2种培养条件下同时检测到的显著性SNP位点
2.6 候选基因预测及表达量分析
依据LD衰减距离,通过中国春基因组数据库,对上述18个显著SNP连锁区间内的序列进行检索和比对,共筛选出12个主效候选基因,利用Ensemble Plants及WheatOmics等网站对候选基因进行功能注释,发现其主要编码MLO蛋白、转运蛋白、解毒蛋白、酰基载体蛋白合成酶和醛脱氢酶等(表6)。
基于expVIP网站获取这12个候选基因在不同组织中的表达信息,这些候选基因均在根系中高表达,根据表达模式可将其分为4类(图8)。第1类包含3个基因(、和),在根系中表达量最高,在茎叶、穗部和籽粒当中表达量均较少;第2类包含4个基因(、、和),在各组织中的表达量为根系>穗部>茎叶>籽粒;第3类包含2个基因,和,在根系和籽粒中表达量较高,茎叶和穗部较少;第4类包含3个基因(、和),在根系当中表达量最高,穗部和籽粒次之,茎叶中几乎不表达。
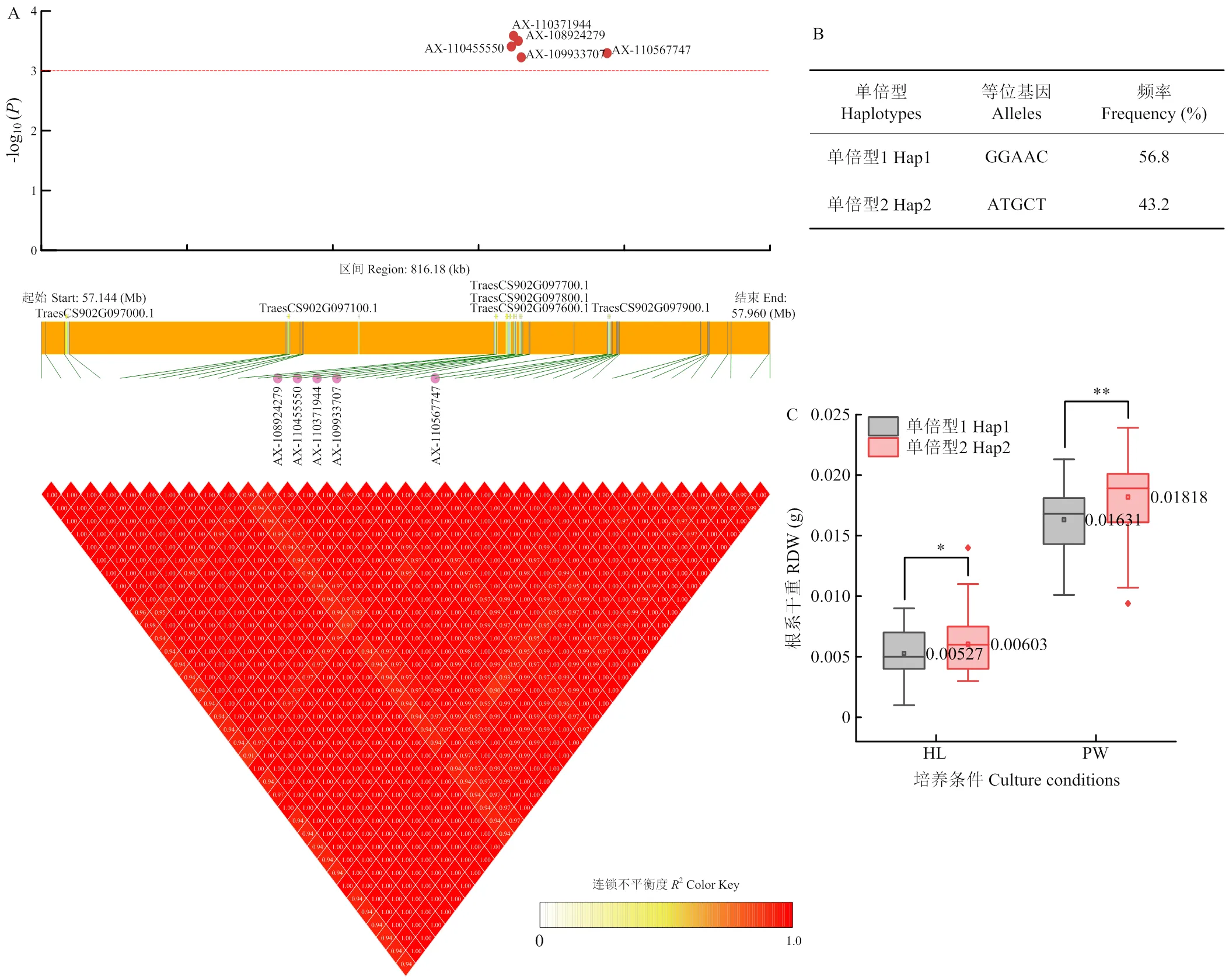
A:2B染色体57.144—57.960 Mb连锁热图;B:不同等位基因的2种单倍型;C:不同单倍型的表型效应
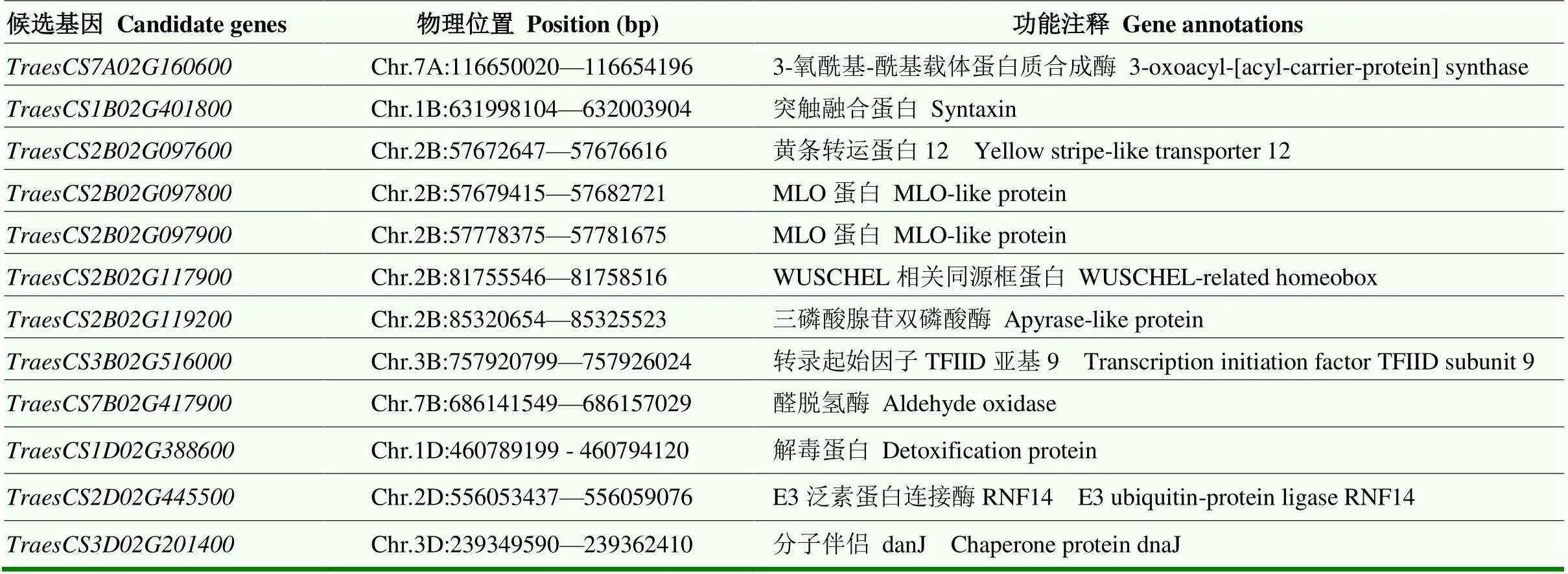
表6 根系相关性状候选基因及其功能注释
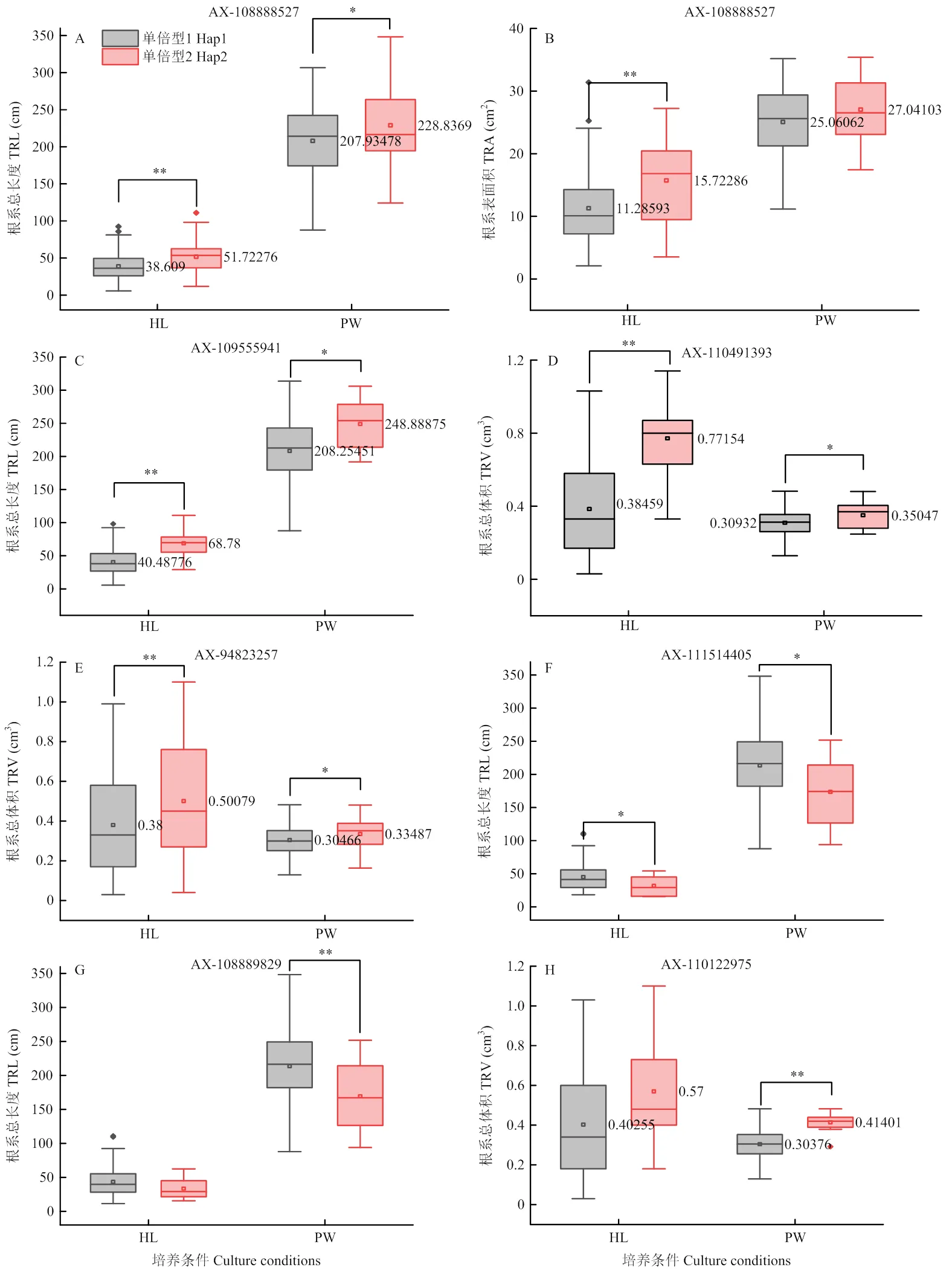
图7 小麦苗期根系性状显著性SNP位点的单倍型分析
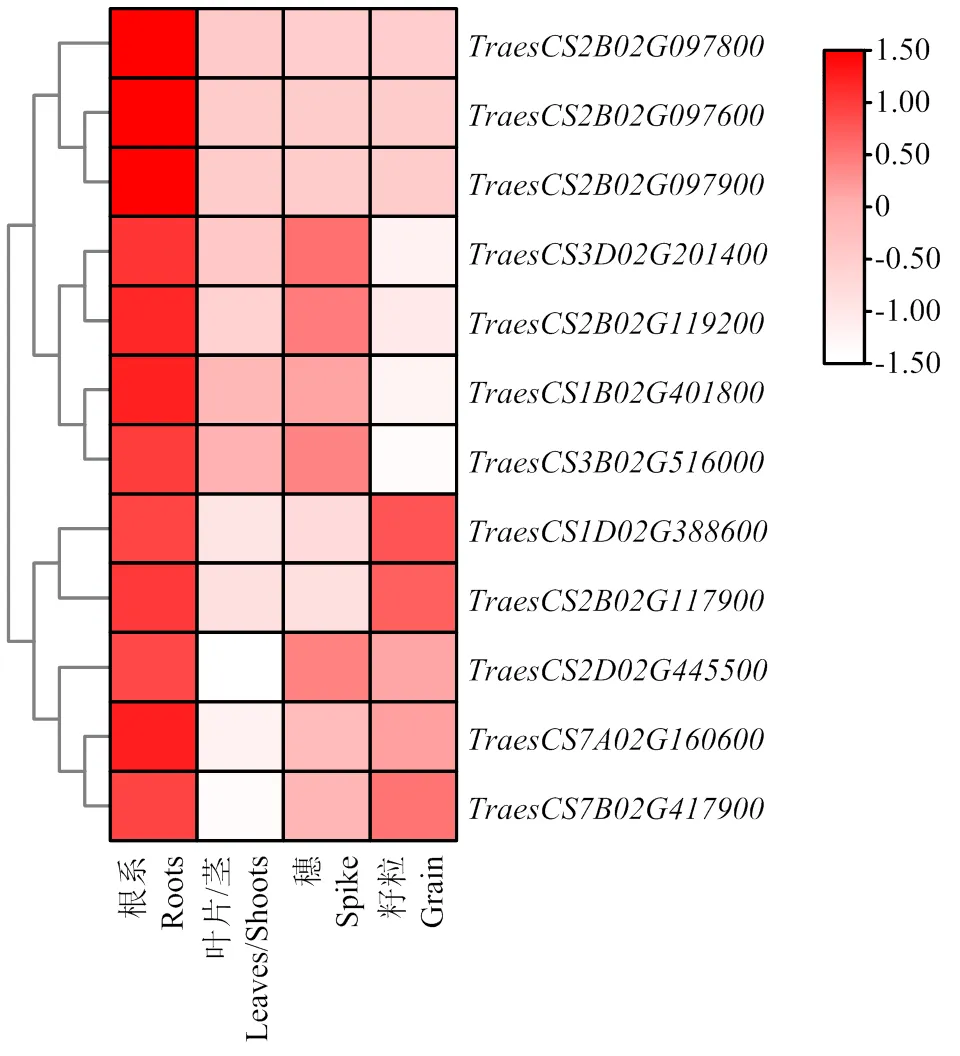
红色代表该基因在某组织中表达量高;粉色或白色则代表在某组织表达量低
2.7 KASP标记开发及小麦自然群体的检测
为了对小麦根系进行分子标记辅助选择,依据2个培养条件下共定位到的显著性SNP设计KASP引物,在根系极端材料中进行初步筛选,最终依据AX-110497143和AX-109933707开发了2个KASP标记(和),对189份小麦品种组成的自然群体进行检测,2个标记能够对其中171份小麦品种进行明显分型,而剩余18份材料分型不明显(图9)。标记鉴定出基因型为CC的小麦品种165份,基因型为TT的6份,两者间的根系总体积差异极显著(=2×10-6);标记鉴定出CC基因型的94份,TT基因型的77份,两者间的根系干重差异显著(=0.001,表7)。
3 讨论
3.1 不同培养条件下小麦苗期根系性状的表型差异
苗期是小麦形态器官建成的关键阶段[29],苗期的生长发育直接关系到后期生长的好坏。而根系作为植物三大器官之一,是植物吸收矿物质养分、水分传导的第一部位,是植物生长发育的源泉,对植物生长起着固定和支持作用。有研究表明[30]小麦成穗数与小麦根系发达程度,特别是和种子根数和根系表面积及体积呈明显的正相关,从而促进小麦产量的提高。在本研究中,2种培养条件下的根系构型具有明显差异:HL培养条件下的根系整体较粗且短;PW培养条件下的主根细而长,侧根也较多。相关性分析表明,各根系性状间具有显著的相关性,说明根系总长度、根系总表面积和总体积等根系性状间可能存在协同互作,有效地提高水分吸收效率[31]。此外,根系长度和根系直径负相关,这可能是造成PW培养条件下根系较长但直径较小的原因。由此推测在PW培养条件下,植株胚乳中的养分足以供给苗期小麦的生长发育,过多的营养可能会抑制幼苗根系的生长。营养元素对小麦植株的生长不可或缺,但施用过多非但无益,反而造成更严重的后果。有研究表明[32],氮素过剩会导致植株生育延迟,易感病虫害;镁元素过剩会阻碍植株生长等。因此,在短期的苗期水培试验中可直接采用去离子水培养,可能取得更佳的效果。
3.2 小麦苗期根系性状显著关联位点分析
本研究对5个小麦苗期的根系性状进行全基因组关联分析,2个培养条件下共检测到95个显著性关联位点,2个条件下均检测到的有18个。其中,4个SNP位点与前人通过不同小麦群体进行全基因组关联分析定位的位点相近或一致。如本研究检测到的4A和5B染色体上与根系干重显著关联的标记AX-109480568和AX-111009146和王博华等[33]利用国内外300份小麦群体定位的根系干重显著关联标记及分别相距6.5和6.0 Mb;1D染色体与根系总长度显著关联的标记AX-111062954和Xu等[34]利用中国黄淮麦区196份小麦材料在2个环境下共定位的根系总长度关联标记相距0.1 Mb;3B染色体与根系总面积显著关联的标记AX-109948487和Liu等[35]利用黄淮麦区165份小麦群体定位的根系总表面积相关标记相同。其余显著关联位点为新发现的,进一步对其进行深入分析可能对解析根系建成遗传机制具有重要意义。
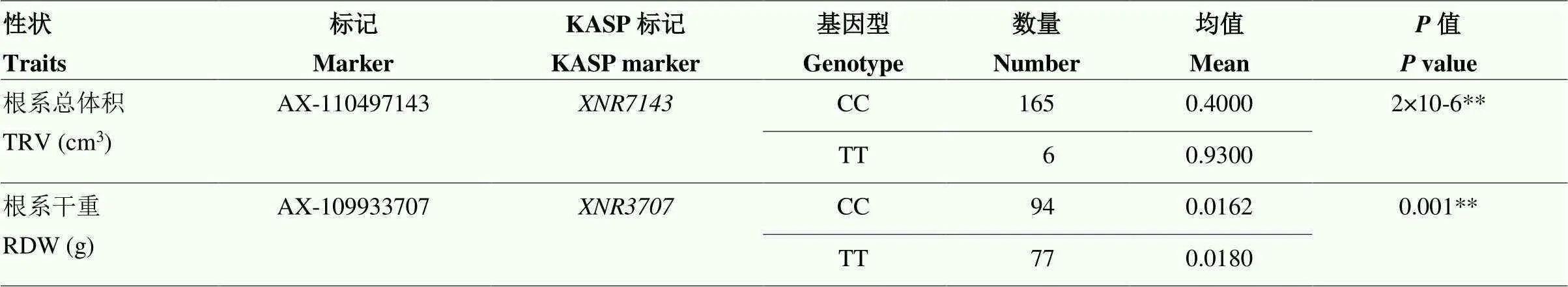
表7 XNR7143和XNR3707在小麦自然群体中的表型验证
**表示在<0.01水平差异显著 ** represent significance of difference at<0.01
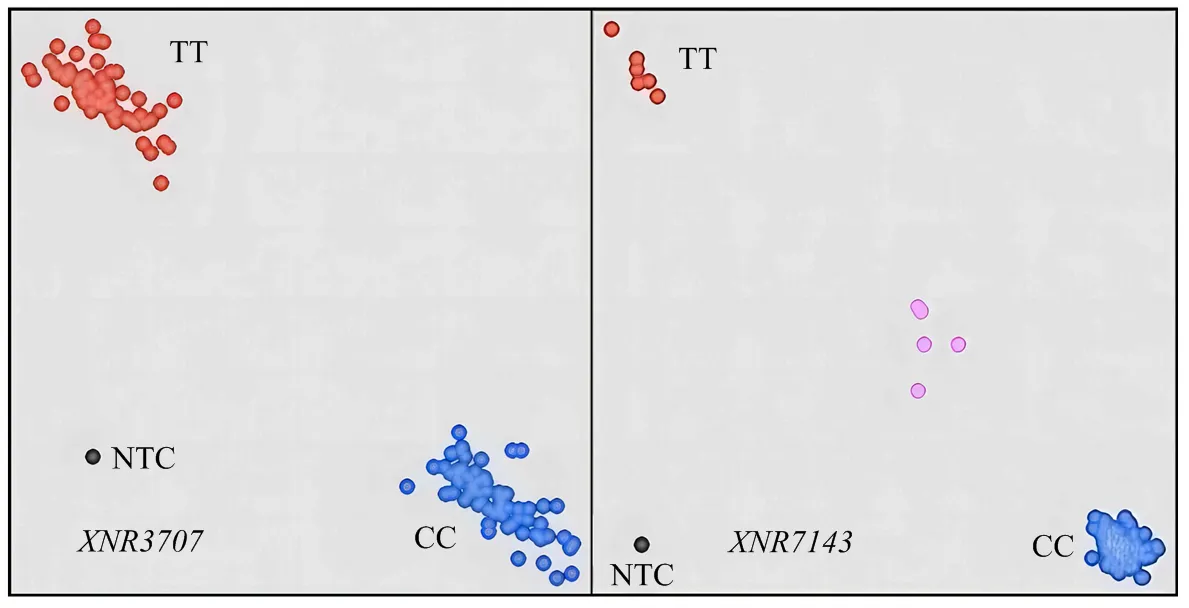
红色代表标记的碱基红色代表标记的碱基为TT的纯合个体;蓝色代表标记的碱基为CC的纯合个体;黑色代表无DNA对照;粉色是无法分型的个体
3.3 小麦根系候选基因功能预测
本研究通过小麦根系性状显著关联的SNP标记序列对小麦中国春基因组序列进行比对分析,依据基因功能注释发现了12个可能调控小麦根系发育的候选基因。其中位于7A染色体的()编码3-氧酰基-酰基载体蛋白合成酶,研究表明该酶参与植物脂肪酸从头合成的缩合反应,在脂肪酸合成过程中扮演重要角色[36]。水稻同源基因过表达,其主根长和侧根明显缩短,根部伸长区和成熟区的细胞长度变短,根中棕榈酸和亚麻酸含量明显增加[37],推测可能是通过参与根系脂肪酸的合成影响小麦根系发育。位于1B染色体的()编码突触融合蛋白,具有SNARE结构域,该蛋白除参与膜泡运输外,还与植物抗病性及植物的向重力性有关[38],推测可能通过参与根冠造粉体的形成,从而保证根系的向地生长。位于2B染色体的()和()编码MLO蛋白,研究表明,植物MLO蛋白除与白粉病菌互作外,还受到干旱胁迫和脱落酸诱导,拟南芥突变会导致其不正常的根部形态,发生严重的卷曲[39]。位于2B染色体的()编码WUSCHEL蛋白,WUSCHEL相关同源盒是植物特异转录因子家族,具有调节植物干细胞分裂分化动态平衡和植物器官发育等重要功能[40]。其水稻同源基因是不定根生长发育的关键调节因子,过量表达使不定根提早产生,数目增加;此外该基因的表达还受外源生长素和细胞分裂素的诱导[41]。位于2B染色体的()编码三磷酸腺苷双磷酸酶,该酶调节胞内外ATP稳态,在植物的各种应激适应及胁迫中发挥重要作用[42]。其水稻同源基因的一个单碱基替换导致异常的cDNA剪接,从而影响水稻根毛细胞的膨大,对水稻根毛伸长及植株生长发育十分重要[43]。位于7B染色体的TraesCS7B02G417900()编码醛脱氢酶,而植物醛脱氢酶是植物激素脱落酸生物合成的最后一步,因此可能通过合成ABA进而调控主根、侧根及根毛的生长发育[44]。位于3D染色体的()编码分子伴侣dnaJ,参与新生肽的折叠、装配和运输过程。研究表明dnaJ-like蛋白在植物形态建成及非生物胁迫方面具有重要作用[45],推测可能通过调节根系发育来响应干旱胁迫、参与重金属解毒。对这些候选基因的进一步功能解析,将有助于构建小麦根系调控的基因网络。
3.4 小麦根系分子标记的开发与检测
KASP技术作为一种基于荧光检测的基因分型技术,具有高通量、低成本、准确性高及减少传统育种工作量等优点,目前,已经广泛应用于植物、动物和人类的遗传学研究与群体分型[46-49]。本研究通过对2种培养条件下共定位的显著性SNP进行KASP标记开发,最终筛选出能够明显分型的和2个KASP标记,其中将171份小麦群体分为根系总体积不同的2个基因型,频率分别为96.5%和3.5%;而将供试材料分为与根系干重相关的2个基因型,频率为55.0%和45.0%。此外,试验表明分型的2个亚群之间的数目具有明显差异,CC基因型为主要亚群,这可能是由于人工选择等因素导致。以上2个分子标记可用作小麦优异种质资源的快速鉴定,为辅助选择优良根系建成的基因型提供了分子标记。
4 结论
不同水培条件下,小麦苗期根系性状存在不同程度的差异:霍格兰营养液培养条件下的根系直径较粗且根长较短;而去离子水培养条件下的主根细长,侧根居多。通过利用660K SNP芯片对小麦苗期的5个根系性状进行全基因组关联分析发现:4个模型同时检测到95个显著关联的SNP位点,其中,在2种培养条件下均检测到的SNP位点有18个,同时基于上述共定位的多态性SNP开发了与根系总体积及根系干重相关的2个KASP标记(和),挖掘到12个可能与根系发育相关的候选基因。
[1] RAY D K, MUELLER N D, WEST P C, FOLEY J A. Yield trends are insufficient to double global crop production by 2050. Plos One, 2013, 8(6): e66428.
[2] SHARMA I, TYAGI B, SINGH G, VENKATESH K, GUPTA O. Enhancing wheat production-A global perspective. Indian Journal of Agricultural Sciences, 2015, 85(1): 3-13.
[3] 郭海宇. 植物根系图像的特征分析方法研究与实现. 电子科技大学计算机软件与理论, 2013.
GUO H Y. Research and implementation of feature analysis method of plant root images. Computer Software and Theory of University of Electronic Science and Technology, 2013. (in Chinese)
[4] CHEN X X, DING Q S, BŁASZKIEWICZ Z, SUN J A, SUN Q, HE R Y, LI Y N. Phenotyping for the dynamics of field wheat root system architecture. Scientific Reports, 2017, 7: 37649.
[5] ATKINSON J A, POUND M P, BENNETT M J, WELLS D M. Uncovering the hidden half of plants using new advances in root phenotyping. Current Opinion in Biotechnology, 2019, 55: 1-8.
[6] LI X K, GUO Z L, LV Y, CEN X, DING X P, WU H, LI X H, HUANG J P, XIONG L Z. Genetic control of the root system in rice under normal and drought stress conditions by genome-wide association study. Plos Genetics, 2017, 13(7): e1006889.
[7] MACCAFERRI M, EL-FEKI W, NAZEMI G, SALVI S, CANÈ M A, COLALONGO M C, STEFANELLI S, TUBEROSA R. Prioritizing quantitative trait loci for root system architecture in tetraploid wheat. Journal of Experimental Botany, 2016, 67(4): 1161-1178.
[8] XIE Q, FERNANDO K M C, MAYES S, SPARKES D L. Identifying seedling root architectural traits associated with yield and yield components in wheat. Annals of botany, 2017, 119(7): 1115-1129.
[9] MAN J G, SHI Y, YU Z W, ZHANG Y L. Dry matter production, photosynthesis of flag leaves and water use in winter wheat are affected by supplemental irrigation in the Huang-Huai-Hai plain of China. Plos One, 2015, 10(9): e0137274.
[10] MA J H, ZHAO D Y, TANG X X, YUAN M, ZHANG D J, XU M Y, DUAN Y Z, REN H Y, ZENG Q D, WU J H, HAN D J, LI T, JIANG L N. Genome-wide association study on root system architecture and identification of candidate genes in wheat (L.). International Journal of Molecular Sciences, 2022, 23(3): 1843.
[11] 陈贵菊, 靳义荣, 刘彩云, 贾德新, 樊庆琦, 刘金栋, 刘鹏. 普通小麦根系建成相关性状的全基因组关联分析. 植物遗传资源学报, 2020, 21(4): 975-983.
CHEN G J, JIN Y R, LIU C Y, JIA D X, FAN Q Q, LIU J D, LIU P. Genome-wide association study of root system architecture related traits in common wheat (L.). Journal of Plant Genetic Resources, 2020, 21(4): 975-983. (in Chinese)
[12] HUANG F, CHEN Z Y, DU D J, GUAN P F, CHAI L L, GUO W L, HU Z R, XIN M M, PENG H R, YAO Y Y, NI Z F. Genome-wide linkage mapping of QTL for root hair length in a Chinese common wheat population. The Crop Journal, 2020, 8(6): 1049-1056.
[13] KHODAEE S M M, HASHEMI M, MIRLOHI A, MAJIDI M M, SUKUMARAN S, ESMAELZAEH MOGHADDAM M, ABDOLLAHI M. Root characteristics of an elite spring wheat panel under contrasting water treatments and their genome-wide association study. Rhizosphere, 2021, 19: 100413.
[14] 杨青青, 唐家琪, 张昌泉, 高继平, 刘巧泉. KASP标记技术在主要农作物中的应用及展望. 生物技术通报, 2022, 38(4): 58-71.
YANG Q Q, TANG J Q, ZHANG C Q, GAO J P, LIU Q Q. Application and prospect of KASP marker technology in main crops. Biotechnology Bulletin, 2022, 38(4): 58-71. (in Chinese)
[15] DELANNAY X, MCLAREN G, RIBAUT J M. Fostering molecular breeding in developing countries. Molecular Breeding, 2012, 29(4): 857-873.
[16] KUMAR S, BANKS T W, CLOUTIER S. SNP discovery through next-generation sequencing and its applications. International Journal of Plant Genomics, 2012, 2012: 831460.
[17] QURESHI N, BARIANA H S, ZHANG P, MCINTOSH R, BANSAL U K, WONG D, HAYDEN M J, DUBCOVSKY J, SHANKAR M. Genetic relationship of stripe rust resistance genesand
[18] FANG T L, LEI L, LI G Q, POWERS C, HUNGER R M, CARVER B F, YAN L L. Development and deployment of KASP markers for multiple alleles of
[19] REHMAN S U, ALI SHER M, SADDIQUE M A B, ALI Z, KHAN M A, MAO X G, IRSHAD A, SAJJAD M, IKRAM R M, NAEEM M, JING R L. Development and exploitation of KASP assays for genes underpinning drought tolerance among wheat cultivars from Pakistan. Frontiers in Genetics, 2021, 12: 684702.
[20] 王志伟, 乔祥梅, 王志龙, 杨金华, 程加省, 程耿, 于亚雄. 云南小麦品种(系)抗逆性相关基因的KASP标记检测. 西南农业学报, 2020, 33(8): 1601-1607.
WANG Z W, QIAO X M, WANG Z L, YANG J H, CHENG J S, CHENG G, YU Y X. Identification of genes associated with stress resistance in Yunnan wheat cultivars (lines) by KASP assays. Southwest China Journal of Agricultural Sciences, 2020, 33(8): 1601-1607. (in Chinese)
[21] BRADBURY P J, ZHANG Z W, KROON D E, CASSTEVENS T M, RAMDOSS Y, BUCKLER E S. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics, 2007, 23(19): 2633-2635.
[22] YIN L L, ZHANG H H, TANG Z S, XU J Y, YIN D, ZHANG Z W, YUAN X H, ZHU M J, ZHAO S H, LI X Y, LIU X L. rMVP: a memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genomics, Proteomics & Bioinformatics, 2021, 19(4): 619-628.
[23] LIU K J, MUSE S V. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics, 2005, 21(9): 2128-2129.
[24] ALEXANDER D H, NOVEMBRE J, LANGE K. Fast model-based estimation of ancestry in unrelated individuals. Genome Research, 2009, 19(9): 1655-1664.
[25] ZHANG C, DONG S S, XU J Y, HE W M, YANG T L. PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics, 2019, 35(10): 1786-1788.
[26] YU S Z, WU J H, WANG M, SHI W M, XIA G M, JIA J Z, KANG Z S, HAN D J. Haplotype variations in QTL for salt tolerance in Chinese wheat accessions identified by marker-based and pedigree- based kinship analyses. The Crop Journal, 2020, 8(6): 1011-1024.
[27] CHEN C J, CHEN H, ZHANG Y, THOMAS H R, FRANK M H, HE Y H, XIA R. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Molecular Plant, 2020, 13(8): 1194-1202.
[28] HAO C Y, WANG L F, GE H M, DONG Y C, ZHANG X Y. Genetic diversity and linkage disequilibrium in Chinese bread wheat (L.) revealed by SSR markers. PlosOne, 2011, 6(2): e17279.
[29] PFLUGFELDER D, KOCHS J, KOLLER R, JAHNKE S, MOHL C, PARIYAR S, FASSBENDER H, NAGEL K A, WATT M, VAN DUSSCHOTEN D. The root system architecture of wheat establishing in soil is associated with varying elongation rates of seminal roots: quantification using 4D magnetic resonance imaging. Journal of Experimental Botany, 2022, 73(7): 2050-2060.
[30] FAN Y, MIGUEZ-MACHO G,JOBBÁGY E G, JACKSON R B, OTERO-CASAL C. Hydrologic regulation of plant rooting depth. Proceedings of the National Academy of Sciences of the United States of America, 2017, 114(40): 10572-10577.
[31] MA S C, XU B C, LI F M, LIU W Z, HUANG Z B. Effects of root pruning on competitive ability and water use efficiency in winter wheat. Field Crops Research, 2008, 105(1/2): 56-63.
[32] 陈德扬. 植物营养元素功能及缺乏和过剩症状(一). 农家之友, 2000(10): 13.
CHEN D Y. Function of plant nutrient elements and symptoms deficiency and excess(Ⅰ). Farmer's Home, 2000(10): 13. (in Chinese)
[33] 王博华, 任毅, 时晓磊, 王继庆, 谢磊, 加娜尔·拜合提, 耿洪伟. 干旱胁迫下小麦苗期根系性状的全基因组关联分析. 植物遗传资源学报, 2022, 23(4): 1111-1123.
WANG B H, REN Y, SHI X L, WANG J Q, XIE L, JIANAER B H T, GENG H W. Genome-wide association analysis of seedling root traits in wheat under drought stress. Journal of Plant Genetic Resources, 2022, 23(4): 1111-1123. (in Chinese)
[34] XU F D, CHEN S L, YANG X W, ZHOU S M, CHEN X, LI J, ZHAN K H, HE D X. Genome-wide association study on seminal and nodal roots of wheat under different growth environments. Frontiers in Plant Science, 2021, 11: 602399.
[35] LIU W B, NI J, SHAH F A, YE K Q, HU H, WANG Q J, WANG D D, YAO Y Y, HUANG S W, HOU J Y, LIU C H, WU L F. Genome-wide identification, characterization and expression pattern analysis offamily members in response to abiotic and biotic stresses in wheat. PeerJ, 2019, 7: e7622.
[36] ABBADI A, BRUMMEL M, SCHÜTT B S, SLABAUGH M B, SCHUCH R, SPENER F. Reaction mechanism of recombinant 3-oxoacyl-(acyl-carrier-protein) synthase III fromembryo, a fatty acid synthase type II condensing enzyme. The Biochemical Journal, 2000, 345(Pt1): 153-160.
[37] DING W N, LIN L, ZHANG B T, XIANG X B, WU J, PAN Z C, ZHU S H. OsKASI, a β-ketoacyl-[acyl carrier protein] synthase I, is involved in root development in rice (L.). Planta, 2015, 242(1): 203-213.
[38] 鲍永美, 王州飞, 张红生. 植物SNARE蛋白的结构与功能. 植物学通报, 2005, 22(6): 715-722.
BAO Y M, WANG Z F, ZHANG H S. Structure and function of SNAREs in plant. Chinese Bulletin of Botany, 2005, 22(6): 715-722. (in Chinese)
[39] 张孝廉, 张吉顺, 雷波, 余婧, 赵德刚. 植物MLO蛋白研究进展. 植物生理学报, 2018, 54(7): 1159-1171.
ZHANG X L, ZHANG J S, LEI B, YU J, ZHAO D G. Research progress of MLO proteins in plants. Plant Physiology Journal, 2018, 54(7): 1159-1171. (in Chinese)
[40] LI Z, LIU D, XIA Y, LI Z L, JING D D, DU J J, NIU N, MA S C, WANG J W, SONG Y L, YANG Z Q, ZHANG G S. Identification of the WUSCHEL-related homeobox () gene family, and interaction and functional analysis ofandin wheat. International Journal of Molecular Sciences, 2020, 21(5): 1581.
[41] ZHAO Y, HU Y F, DAI M Q, HUANG L M, ZHOU D X. The WUSCHEL-related homeobox geneis required to activate shoot-borne crown root development in rice. The Plant cell, 2009, 21(3): 736-748.
[42] 刘文博. 小麦APYRASE家族成员鉴定及功能研究[D]. 合肥: 中国科学技术大学, 2019.
LIU W B. Identification and characterization of wheatgene family members[D]. Hefei: University of Science and Technology of China, 2019. (in Chinese)
[43] YUO T, TOYOTA M, ICHII M, TAKETA S. Molecular cloning of a root hairless gene
[44] ZAREPOUR M, SIMON K, WILCH M, NIELÄNDER U, KOSHIBA T, SEO M, LINDEL T, BITTNER F. Identification of superoxide production byaldehyde oxidases AAO1 and AAO3. Plant Molecular Biology, 2012, 80(6): 659-671.
[45] 黄守程, 叶梅荣, 刘爱荣, 张远兵. DnaJ-like蛋白在植物胁迫应答中的作用及机制研究进展. 长江大学学报(自科版), 2015, 12(9): 57-62.
HUANG S C, YE M R, LIU A R, ZHANG Y B. Research progress on the role and mechanism of DnaJ-like protein in plant stress response. Journal of Yangtze University (natural science edition), 2015, 12(9): 57-62. (in Chinese)
[46] CHANG C C, SILVA B B I, HUANG H Y, TSAI C Y, FLORES R J D, TAYO L L, TYAN Y C, TSAI M A, CATULIN G E M, CHUANG K P, YANG J L. Development and validation of KASP assays for the genotyping of racing performance-associated single nucleotide polymorphisms in pigeons. Genes, 2021, 12(9): 1383.
[47] AYALEW H, TSANG P W, CHU C G, WANG J Z, LIU S Y, CHEN C F, MA X F. Comparison of TaqMan, KASP and rhAmp SNP genotyping platforms in hexaploid wheat. Plos One, 2019, 14(5): e0217222.
[48] LANDOULSI Z, BENROMDHAN S, BEN DJEBARA M, DAMAK M, DALLALI H, KEFI R, ABDELHAK S, GARGOURI-BERRECHID A, MHIRI C, GOUIDER R. Using KASP technique to screen
[49] HIREMATH P J, KUMAR A, PENMETSA R V, FARMER A, SCHLUETER J A, CHAMARTHI S K, WHALEY A M, CARRASQUILLA-GARCIA N, GAUR P M, UPADHYAYA H D, KAVI KISHOR P B, SHAH T M, COOK D R, VARSHNEY R K. Large-scale development of cost-effective SNP marker assays for diversity assessment and genetic mapping in chickpea and comparative mapping in legumes. Plant Biotechnology Journal, 2012, 10(6): 716-732.
Genome-Wide Association Studies and Mining for Favorable Loci of Root Traits at Seedling Stage in Wheat
WANG Mai1, DONG QingFeng1, GAO ShenAo1, LIU DeZheng1, LU Shan1, QIAO PengFang1, CHEN Liang1, HU YinGang1,2
1College of Agronomy, Northwest A & F University/State Key Laboratory of Crop Stress Biology for Arid Areas, Yangling 712100, Shaanxi;2Institute of Water-Saving Agriculture in Arid Areas of China, Yangling 712100, Shaanxi
【Objective】Plant roots are critical for water and nutrient acquisition, crop growth and development as well as yield formation. Exploring SNP loci significantly associated with root traits in wheat at seedling stage and mining candidate genes, will lay a foundation for understanding the genetic mechanism of wheat root system architecture and breeding wheat elite varieties with better root architecture.【Method】In this study, 189 diverse wheat cultivars were assembled as an association-mapping panel, five root traits including total root length (TRL), total root area (TRA), total root volume (TRV), average root diameter (ARD) and root dry weight (RDW) were investigated by growing in two culture conditions (Hoagland nutrient solution and pure water), and the experiments were repeated twice. Then, genome-wide association studies (GWAS) were performed for the five root traits with genotypic data derived from Wheat 660K SNP Array. Candidate genes were predicted by sequence alignment, domain analysis, and annotation information. Futhermore, kompetitive allele specific PCR (KASP) markers were developed for root traits. 【Result】The root traits varied greatly among the 189 cultivars, and the roots were thick and short cultured under Hoagland nutrient solution, while slender seminal roots and more lateral roots were observed under pure water. A total of 95 QTLs significantly associated with root traits cultured in two conditions (<10-3) were identified by genome-wide association studies with four models of BLINK (bayesian-information and linkage-disequilibrium iteratively nested keyway), CMLM (compressed mixed linear model), FarmCPU (fixed and random model circulating probability unification) and MLM (mixed linear model). Among them, 18 QTLs were detected in both culture conditions and distributed on chromosomes of 7A, 1B, 2B, 3B, 7B, 1D, 2D, and 3D, which explained 8.68%-14.07% of phenotypic variation. Of those significant loci, 4 QTLs were similar or consistent with thatreported previously, and the rest were novel ones. Haplotype analysis conducted for co-localization QTLs of 10 SNPs revealed significant differences in root traits between the two haplotypes of wheat cultivars. Based on these SNPs, KASP markersandwere developed for total root volume and root dry weight, respectively. In addition, 12 candidate genes possibly regulating root development were found by mining the genes within the interval of co-localization significant SNPs. Of them,, encoding 3-oxoacyl-[acyl-carrier-protein] synthase, is involved in the synthesis of root fatty acids;,encoding syntaxin, plays an important role in plant tropism;, encoding aldehyde oxidase, contributes to the synthesis of abscisic acid and regulation of crop root development. 【Conclusion】The root traits of wheat varied significantly among the wheat genotypes. Genome-wide association studies detected 18 significant QTLs linked with root traits simultaneously in two culture conditions, two KASP markers were developed for root traits, and 12 candidate genes related to root development were screened, which might provide reference for understanding the regulation mechanism of wheat root traits and molecular marker-assisted breeding for wheat improvement.
wheat; root traits; genome-wide association study; co-localization SNPs; KASP (kompetitive allele specific PCR) markers; candidate genes

10.3864/j.issn.0578-1752.2023.05.001
2022-10-06;
2022-12-13
陕西省重点研发计划(2021KWZ-23)
王脉,E-mail:599157540@qq.com。通信作者胡银岗,E-mail:huyingang@nwafu.edu.cn
(责任编辑 李莉)
