改性粒状硫酸钙制备及其在聚氯乙烯中的应用
2023-03-12房可能毕永香陈前林
徐 丽,房可能,毕永香,杨 敏,陈前林
(贵州大学化学与化工学院,贵州贵阳 550025)
磷石膏(PG)是湿法磷酸生产过程中产生的固体废弃物,主要成分为CaSO4·2H2O。此外,磷石膏中还含有可溶性磷、氟、金属盐类和磷矿浮选残余的有机物等杂质,其早期处理方式以堆存为主[1-2]。2020年中国PG产量约为75 Mt,利用率仅为45.3%[3]。PG大量堆存不仅浪费资源,还造成土壤、地下水和大气等环境污染。因此,PG的资源化和高值化利用是磷化工行业的研究热点之一。
利用PG制备石膏制品是其高附加值利用的途径之一。根据含水量的不同,石膏制品常分为二水硫酸钙(CaSO4·2H2O)、半水硫酸钙(CaSO4·0.5H2O)和无水硫酸钙(CaSO4)三相[4]。CaSO4·2H2O晶体内部含有水通道,在应用过程中会因为失去结晶水造成晶体缺陷,导致其性能不稳定;CaSO4·0.5H2O容易发生水化而改变性质;CaSO4结构紧密,性能稳定不易水化,且力学性能优异[5-6]。目前,CaSO4的制备方法主要有一步水热法、水热-焙烧法和常压酸化法。一步水热法制备无水硫酸钙的要求苛刻、产量小并且能耗高,难以满足大规模工业生产[7]。传统水热-焙烧法是先采用水热法制备出CaSO4·0.5H2O,再通过高温焙烧脱水转化成CaSO4,但是焙烧需要强行从CaSO4·0.5H2O晶格中脱去半个H2O分子,会导致晶体结构崩塌[8]。常压酸化法是将高浓度的二水石膏浑浊液置于酸性溶液中转化成CaSO4,无需高温高压就能实现一步转化[9]。吕智慧等[5]采用常压酸化法将生石膏溶在浓盐酸与水的混合溶液中,于70 ℃反应4 h,制得长为10~20 μm、直径为0.1~0.3 μm的CaSO4晶须。谢晴等[10]采用常压酸化法,以固液质量体积比为0.10 g/mL将磷石膏溶在盐酸中,在90 ℃反应3 h后成功制得长径比为3~8的CaSO4晶须。
CaSO4在结晶过程中具有原子排列高度有序、晶体结构完整、热稳定性好且强度高等特点[11]。但是,长径比大的CaSO4晶须单独使用时表面自由能高,将其应用于聚合物中时会因为高温失去结晶水进而使其长径比变短,表面自由能降低,导致与高分子聚合物相容性差,最终失去增强增韧的效果。鉴于此,制备非晶须状的无水微米硫酸钙(粒状硫酸钙,G-CaSO4),不仅保留了普通粉体硫酸钙的轻质、胶凝性和阻燃性好的优良性能,而且作为塑料填料具有结构完整、高模量、高抗拉强度且易进行表面处理等特性[12-13],还可以在很大程度上提高塑料的综合机械性能。
聚氯乙烯(PVC)作为三大通用塑料之一,由于其抗冲击强度、抗微裂纹扩展能力低等缺陷限制了其广泛应用。因此,笔者使用常压酸化法以磷石膏为原料制备得到粒状硫酸钙(G-CaSO4),再使用硬脂酸将G-CaSO4表面改性制得改性粒状硫酸钙(MG-CaSO4),将MG-CaSO4作为填料应用在PVC中,旨在增强复合材料的力学性能,同时实现磷石膏的高值化利用。
1 实验部分
1.1 原料、试剂和仪器设备
实验原料:磷石膏来自贵州瓮安某化工厂,含水率(质量分数)约为18.14%,呈灰白色,实验前用水洗涤3次除去浆料表面漂浮的有机物,然后置于60 ℃鼓风干燥箱中干燥。干燥磷石膏的主要化学组成见表1。从表1看出,磷石膏主要含有Ca、S、O元素。图1为磷石膏的XRD谱图和SEM照片。从图1a可知,磷石膏主要成分与CaSO4·2H2O标准卡片一致(PDF#33-0311),说明磷石膏的主要成分为CaSO4·2H2O,还含有少量的CaSO4·0.5H2O和SiO2等。从1b看出,磷石膏晶形为不规则的板状,分布松散。其他原料和试剂:SG-5型PVC与PVC加工助剂(316型有机锡热稳定剂、HB-600型润滑剂、H110型PE蜡和氯化聚乙烯);邻苯二甲酸二辛酯(DOP,纯度为99%);硫酸(H2SO4,纯度为96%~98%);无水乙醇(C2H5OH,纯度为99.5%);硬脂酸(分析纯);实验用水均为去离子水。

表1 磷石膏主要化学组成Table 1 Main chemical compositions of PG

图1 磷石膏XRD谱图(a)和SEM照片(b)Fig.1 XRD pattern(a) and SEM image of PG(b)
实验仪器和设备:101-1AB型电热鼓风干燥箱;FA-1004N型电子天平;DF-101S型集热式恒温加热磁力搅拌器;SHZ-D(Ⅲ)型循环水真空泵;YL-200B型超纯水机;XSM-500型密炼机;PC-250型破碎机;PL860型注塑机。
1.2 实验方法
1.2.1 MG-CaSO4制备
采用常压酸化法先制备得到粒状硫酸钙(GCaSO4),再将其表面改性制备得到改性粒状硫酸钙(MG-CaSO4)。具体操作步骤:1)将干燥的磷石膏以液固质量比为3∶1加入到装有硫酸溶液体系的三口烧瓶(已装上温度计和冷凝管)中,开启磁力搅拌(160 r/min)并加热反应一段时间,反应结束后趁热抽滤,滤饼用饱和Ca(OH)2上清液洗涤至碱性(pH=13)后在80 ℃干燥箱中干燥24 h,制得粒状CaSO4(记为G-CaSO4);2)将5%硬脂酸(以G-CaSO4质量计)和100 g无水乙醇加入到三口烧瓶(装有温度计和冷凝管)中,待硬脂酸溶解后加入20 g G-CaSO4,开启磁力搅拌(300 r/min),在60 ℃加热反应2 h,反应结束后趁热抽滤,滤饼用无水乙醇和热水交替洗涤后在80 ℃鼓风干燥箱中干燥24 h,制得硬脂酸改性的G-CaSO4(记为MG-CaSO4)。
1.2.2 PVC复合材料制备
将PVC与PVC加工助剂(5%有机锡热稳定剂、0.6%润滑剂、0.6%PE蜡和4%氯化聚乙烯,以PVC质量计)预混合,分别与G-CaSO4和MG-CaSO4(添加量分别为0、2%、4%、6%、8%、10%,以质量分数计)混合,然后将混合物料与20%(质量分数)邻苯二甲酸二辛酯加入到密炼机中,在180 ℃熔融共混10 min,人工造粒后用破碎机破碎,最后用注塑机注塑成样条(即PVC复合材料),用于力学性能检测。
1.2.3 材料表征与性能测试
使用Ultima IV型X射线衍射仪对样品进行物相分析,确定CaSO4的物相组成。使用∑IGMA型扫描电镜分析CaSO4和材料拉伸断裂面的微观形貌。使用Nicolet is5型红外光谱仪、Tecnai G2-F20型透射电镜(TEM、HR-TEM)进一步分析G-CaSO4的官能团、物相组成和微观结构。使用Nicolet is5型红外光谱仪表征MG-CaSO4的基团。使用TSE-104B型微机控制电子万能试验机进行PVC复合材料的拉伸和弯曲实验。先用XQZ-Ⅱ型电动缺口制样机制样,再通过XJUD-5.5型悬臂梁进行冲击实验测试。
2 结果与讨论
2.1 MG-CaSO4制备
2.1.1 反应温度对G-CaSO4物相组成及形貌的影响
将干燥的磷石膏以液固质量比为3∶1加入到装有硫酸溶液的体系中,固定反应时间为4 h,探究反应温度对产物物相组成和形貌的影响,结果见图2和图3。图2为不同温度制备的产物XRD谱图和(020)面XRD谱图放大图。由图2a可知,90、100、110 ℃制备的产物XRD峰与标准卡片CaSO4(PDF#37-1496)衍射特征峰位置基本吻合,并且在2θ为25.43、31.36、38.64、40.81°处的特征峰分别对应CaSO4的(020)(012)(202)(212)晶面[14],说明制得的产物主要为CaSO4。此外,将(020)特征峰放大后发现,随着温度的升高,特征峰向小角度偏移。这可能是由于温度的升高使得原子之间的运动加剧,进一步使制得的CaSO4晶格间距变大,从而使衍射峰向低角度偏移。
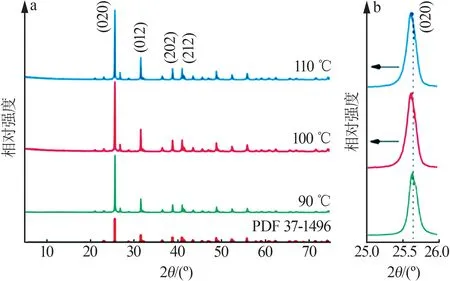
图2 不同温度制备的产物XRD谱图(a)和(020)面XRD谱图放大图(b)Fig.2 XRD patterns of samples prepared at different tempera⁃tures(a) and XRD enlarged patterns of (020) plane(b)
通过MDI Jade 6分析得到不同温度制得的产物半峰宽(FWHM),由FWHM分析产物的结晶性能,数据列于表2。从表2看出,在相同测试条件下,随着反应温度升高,CaSO4各个晶面FWHM均逐渐减小,说明制得的CaSO4结晶状态逐渐稳定,表明温度的升高有利于产物结晶[15]。
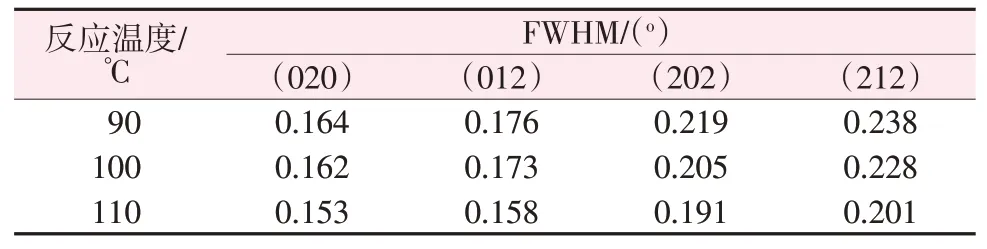
表2 不同温度制得的产物FWHMTable 2 FWHM of samples prepared at different temperatures
图3为不同温度制备的产物SEM照片。从图3可知,反应温度对产物形貌的影响较大。反应温度为90 ℃时,制得的CaSO4呈薄片状。这是因为,温度低时体系中活化分子数较少,磷石膏溶解速率慢,CaSO4成核率低,导致晶体生长速度慢。调节反应温度至100 ℃时,制得的CaSO4呈现均匀的粒状结构。这是因为,磷石膏溶解速率加快,CaSO4成核率增大的同时晶体生长速度快,从而实现完整生长。调节温度至110 ℃(高于H2SO4-H2O的共沸点)时,由于温度过高,增加了CaSO4·2H2O之间的碰撞几率,原子易侧向结合形成板状CaSO4,这与图2的分析结果一致。综上所述,在100 ℃反应4 h可制得形貌较规整且分布均匀的CaSO4。

图3 不同温度制备的产物SEM照片Fig.3 SEM images of samples prepared at different temperatures
2.1.2 反应时间对G-CaSO4物相组成及形貌的影响
固定反应温度为100 ℃,探究反应时间对产物物相组成和形貌的影响,结果见图4和图5。图4为100 ℃不同反应时间制得的产物XRD谱图。从图4a可知,所有产物结构均与Bmmb(63)空间群结构(CaSO4,PDF#37-1496)相符,在2θ为25.43、31.36、38.64、40.81°处的衍射峰分别对应于CaSO4的(020)(012)(202)(212)晶面,说明制得的产物主要为CaSO4。并且随着反应时间的增加,产物的(020)面衍射峰从25.53°移动到25.68°(见图4b),说明反应时间延长,CaSO4晶格收缩导致晶面间距减小,从而使得(020)衍射峰向高角度偏移。
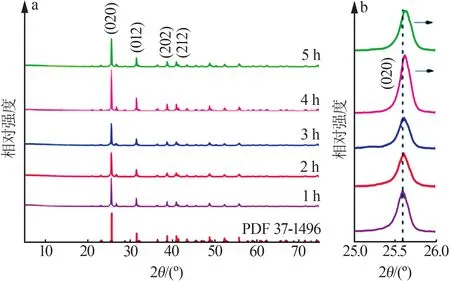
图4 不同反应时间制备的产物XRD谱图(a)和(020)面XRD谱图放大图(b)Fig.4 XRD patterns of samples prepared at different times(a)and XRD enlarged patterns of (020) plane(b)
通过MDI Jade 6分析得到不同反应时间制得的产物半峰宽(FWHM),由FWHM分析产物的结晶性能,数据列于表3。由表3可知,随着反应时间的增加,产物各个晶面FWHM先减小后增大,反应时间为4 h时制得的产物FWHM最小,说明此时制得的CaSO4结晶性能最好。
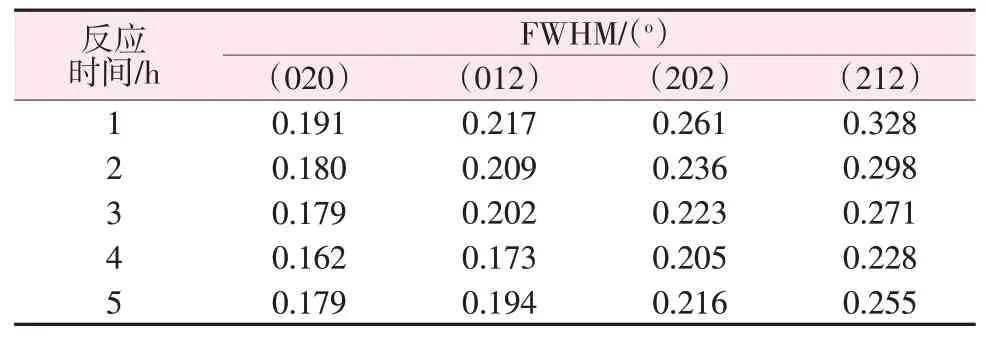
表3 不同反应时间制得的产物FWHMTable 3 FWHM of samples prepared at different times
图5为不同反应时间制得的产物SEM照片。从图5可知,反应时间对产物形貌的影响较小。反应时间为1 h 时,制得的CaSO4聚集在一起。延长反应时间到2~3 h,CaSO4逐渐变规整,但是分布仍然不均匀,且出现板状结构。当反应时间为4~5 h时,CaSO4变成分布均匀、形貌规整且表面光滑的粒状结构(粒状CaSO4)。这是因为,CaSO4的形成要经历CaSO4·2H2O溶解—溶液过饱和—CaSO4晶核形成—晶体生长等一系列过程。在反应的初始阶段,磷石膏溶解不充分。随着时间的延长,溶液中的Ca2+和SO42-达到过饱和,形成少量CaSO4晶核,然后结晶速率逐渐增大,CaSO4晶体开始析出。随着CaSO4晶体析出量增加,溶液中的Ca2+和SO42-不断运动导致能量在固-液界面不断转移,当能量达到动态平衡后,晶体最终实现完全生长[16]。由图5d、e可知,4 h和5 h制得的CaSO4形貌无明显变化。考虑到能耗及CaSO4结晶性能,选择4 h作为制备粒状CaSO4(G-CaSO4)的最佳反应时间。利用 Nano measurer 1.2软件测量G-CaSO4的 SEM照片(图5d),其粒径数据分布如图5f所示,计算得到G-CaSO4的平均粒径为0.27 μm。综上所述,在100 ℃反应4 h成功制得了分布均匀、形貌规整的G-CaSO4。

图5 不同反应时间制得的产物SEM照片(a~e)及G-CaSO4的粒径分布图(f)Fig.5 SEM images of samples prepared at different times(a~e) and particle size distribution of G-CaSO4(f)
2.1.3 最佳条件制备的G-CaSO4官能团、物相和微观形貌分析
对100 ℃反应4 h制得的G-CaSO4进行FT-IR、TEM、HRTEM表征,分析其官能团、物相和微观结构,结果见图6。图6a为PG和G-CaSO4的FT-IR图。由图6a可知,PG样品在波数为3 550、3 408、1 688、1 621 cm-1处分别出现PG中结晶水的羟基伸缩振动吸收峰和弯曲振动吸收峰[17],在1 125 cm-1处出现SO42-的对称伸缩振动峰,在669 cm-1和601 cm-1处出现SO42-的非对称弯曲振动峰[18-19]。经过常压酸化处理,G-CaSO4样品中结晶水的羟基伸缩振动吸收峰和弯曲振动吸收峰消失;G-CaSO4样品在3 458 cm-1处出现—OH峰,可能是样品表面吸附空气中的水形成的,说明PG失去结晶水变成了CaSO4。此外,G-CaSO4样品在1 201 cm-1和1 103 cm-1处的特征吸收峰分别是SO42-的非对称伸缩振动峰和对称伸缩振动峰,在678 cm-1和594 cm-1处的峰为SO42-的非对称弯曲振动峰。综上分析,说明采用PG成功制得了G-CaSO4。

图6 G-CaSO4的FT-IR图(a)、TEM照片(b)和HR-TEM照片(c)Fig.6 FT-IR spectra(a),TEM image(b) and HRTEM image of G-CaSO4(c)
由G-CaSO4的TEM照片(图6b)可知,样品的粒径为微米级。由G-CaSO4的HR-TEM照片(图6c)可知,样品的晶格条纹间距为0.30 nm和0.33 nm,分别对应于G-CaSO4的(002)和(020)晶面间距,与前述XRD分析结果一致。基于上述分析可知,采用常压酸化法成功制得了形貌规整、分布均匀的G-CaSO4,这为其在PVC中的应用奠定了良好基础。
2.1.4 G-CaSO4改性
G-CaSO4是亲水性填料,因表面能不同在聚氯乙烯(PVC)中容易发生团聚,从而造成PVC复合材料力学性能的降低,所以使用5%硬脂酸对其进行表面疏水改性,提高其在PVC中的分散性。为证明硬脂酸成功改性G-CaSO4,对硬脂酸、G-CaSO4和MG-CaSO4进行了FT-IR分析,结果见图7。由图7可知,MG-CaSO4在波数为2 925 cm-1和2 853 cm-1处出现了CH3、CH2的C—H键反对称伸缩振动峰和对称伸缩振动峰,同时在1 420 cm-1处出现羧酸盐的不对称伸缩振动峰,说明MG-CaSO4表面有Ca—O—(O=C)—R生成。这是由于硬脂酸在少量水存在时发生电离生成CH3(CH2)16COO-与G-CaSO4发生微弱电离产生的Ca2+结合,生成疏水性更强的硬脂酸钙,从而包覆在G-CaSO4表面达到疏水改性的效果。在靠近G-CaSO4表面一端的Ca2+由于化学键作用接上硬脂酸钙的亲水基团,同时疏水基团朝向外侧,最终达到对G-CaSO4表面疏水改性的目的[20-22]。由上述结果分析可知,使用5%硬脂酸对G-CaSO4进行疏水改性,成功制得了MG-CaSO4。
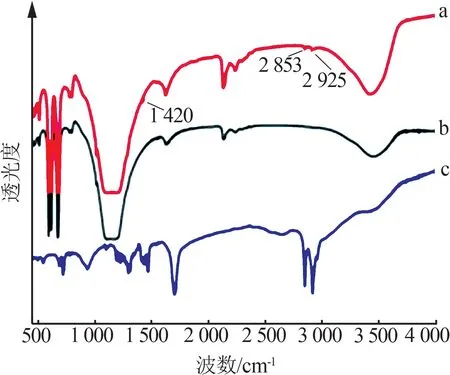
图7 MG-CaSO4(a)、G-CaSO4(b)、硬脂酸(c)的FT-IR图Fig.7 FT-IR spectra of MG-CaSO4(a),G-CaSO4(b)and stearic acid(c)
2.2 MG-CaSO4在PVC中的应用
2.2.1 MG-CaSO4对PVC力学性能的影响
分别将G-CaSO4和MG-CaSO4作为填料添加到PVC中制备得到复合材料,研究了G-CaSO4和MGCaSO4不同添加量对复合材料力学性能的影响,结果见图8。由图8a可知,随着填料添加量从0增加到10%,G-CaSO4/PVC复合材料的拉伸强度呈现下降趋势,而MG-CaSO4/PVC复合材料的拉伸强度呈现先增后降的趋势,在填料添加量为2%时达到最大。说明G-CaSO4添加量的增加可能使填料在PVC基体中分散不均且团聚显著,从而导致填料与基体之间的界面黏合弱,只有少量应力从基体转移到填料表面,进而复合材料横截面的承载能力降低,最终导致PVC复合材料抗拉强度下降[23-25]。改性后,MG-CaSO4与PVC基体界面黏合增强,受到拉伸时难以从基体中脱落,消耗了部分能量,在相同添加量下,MG-CaSO4/PVC复合材料比G-CaSO4/PVC复合材料表现出更好的拉伸性能,添加2%的MGCaSO4,PVC复合材料的拉伸强度提高了11.39%。
由图8b可知,随着填料添加量从0增加到10%,G-CaSO4/PVC复合材料与MG-CaSO4/PVC复合材料的冲击强度呈现增加趋势。这是因为,复合材料受到冲击时倾向于沿着或穿过填料产生裂纹,裂纹扩展路径增加会消耗更多的能量,且填料强度比PVC基体强度高也会消耗能量,所以复合材料的冲击强度得到提高[26]。在相同添加量下,G-CaSO4/PVC复合材料比MG-CaSO4/PVC复合材料表现出更好的抗冲击性能,这是由于分散后MG-CaSO4留在PVC基体中的CaSO4含量比G-CaSO4的少,所以冲击强度相对较弱。
由图8c可知,随着填料添加量从0增加到10%,G-CaSO4/PVC复合材料弯曲强度逐渐减小,而MGCaSO4/PVC复合材料弯曲强度先增大后减小,在填料添加量为4%时达到最大。在相同添加量下,MG-CaSO4/PVC复合材料比G-CaSO4/PVC复合材料表现出更好的弯曲性能,在MG-CaSO4添加量为4%时,PVC复合材料弯曲强度提高了28.50%,其增强机理与拉伸性能一致。
由图8d可知,随着填料添加量从0增加到10%,G-CaSO4/PVC复合材料与MG-CaSO4/PVC复合材料的断裂伸长率均呈现先增大后减小的趋势,在填料添加量为8%时均达到最大。在相同添加量条件下,MG-CaSO4/PVC复合材料比G-CaSO4/PVC复合材料具有更高的断裂伸长率。添加8%的MGCaSO4时,PVC复合材料断裂伸长率提高了51.53%。断裂伸长率的显著变化是因为MG-CaSO4表面具有硬脂酸的柔性烷基链,使得韧性增强[26]。
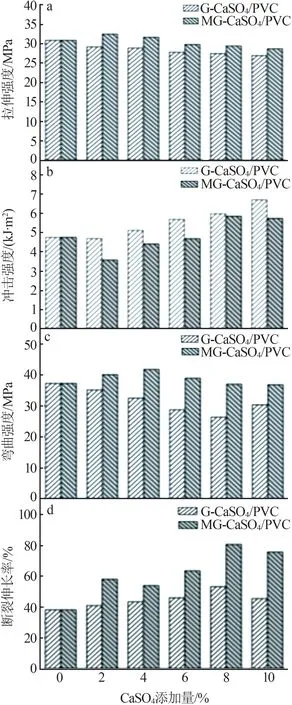
图8 PVC复合材料的力学性能Fig.8 Mechanical properties of PVC composites
2.2.2 MG-CaSO4对PVC复合材料微观形貌的影响
为了分析填料在PVC基体中的分散状态及两者的界面结合情况,采用SEM对MG-CaSO4/PVC拉伸断裂面进行表征,结果见图9。从图9可知:纯PVC表面光滑;分别添加G-CaSO4和MG-CaSO4后,PVC复合材料表面变得粗糙,表明填料的加入使PVC基体发生了变形[27]。从图9b、d看出,当填料添加量为2%时,与G-CaSO4相比,MG-CaSO4能均匀分散并嵌入PVC基体中,证明MG-CaSO4和基体之间具有良好的界面黏合作用,应力能够更好地从基体转移到MG-CaSO4上,从而增强了复合材料的拉伸性能。当G-CaSO4和MG-CaSO4添加量分别从2%变为8%时,G-CaSO4发生了严重团聚(见图9c),且MG-CaSO4有脱黏现象(见图9e),表明在较高含量下填料与PVC基体之间的界面相互作用差,不利于改善复合材料的拉伸性能。综上所述,低含量的MG-CaSO4与PVC基体具有良好的相容性,在其添加量为2%时,复合材料的拉伸强度、断裂伸长率和弯曲强度分别提高了11.39%、41.73%和14.02%,有效地增强了PVC材料的力学性能。

图9 PVC复合材料拉伸断裂面微观形貌Fig.9 Micromorphology of tensile fracture surface of PVC composites
3 结论
以磷石膏为原料,采用常压酸化法制备了GCaSO4。FT-IR、TEM和HRTEM分析表明,制备的G-CaSO4形貌规整、粒度分布均匀、平均粒径为0.27 μm。采用硬脂酸作为改性剂对G-CaSO4进行表面疏水改性获得MG-CaSO4,将MG-CaSO4作为填料应用在PVC中,结果表明MG-CaSO4与PVC基体的界面结合较好,MG-CaSO4能均匀分散并嵌入PVC基体中,起到良好的增强效果。当添加2%的MG-CaSO4时,PVC复合材料的拉伸强度、断裂伸长率和弯曲强度分别提高了11.39%、41.73%和14.02%。因此,本研究通过磷石膏制备得到MG-CaSO4并将其作为填料添加到PVC中,不仅增强了复合材料的力学性能,还能实现磷石膏的高值化利用。