基于msiPL模型的MSI数据分析
2023-03-08张敏,黄钢
张 敏,黄 钢
(1.上海理工大学 健康科学与工程学院,上海 200093;2.上海健康医学院附属嘉定中心医院 上海市分子影像学重点实验室,上海 201318)
0 引言
质谱成像(Mass Spectrometry Imaging,MSI)是一项快速发展的免标记分子成像技术,能够实现蛋白质、肽、脂质、代谢物和药物分子等多种分子的空间分布可视化和相对定量分析,具有高灵敏度和分子特异性[1]。这些分子数据可以在通路识别、生物标志物发现、手术指导、改善临床诊断和预后等方面发挥重要作用[2-4]。近年来,研究者更多关注MSI 技术的空间分辨率、分子覆盖深度和采集吞吐量的提升,大大增加了MSI 的数据量。因此,更有效、更准确地挖掘MSI 数据以识别具有临床重要性的分子特征并实现新的生物标志物发现的计算发展是扩大MSI 应用的有效方式[5]。然而,由于MSI 数据的复杂性,传统的机器学习算法在数据挖掘、聚类、可视化和分类上往往会受到计算机内存和计算速度的限制[6]。原始的高分辨率MSI数据文件可高达数个TB 大小,其中包含了数万个光谱,每个光谱含104~106个质荷比(mass-to-charge ratio,m/z),传统的机器学习算法将造成“维度灾难”。
峰值拾取是分析原始MSI 数据的预处理步骤。峰值拾取可以减轻稀疏性并降低原始光谱维数,同时通过保留尽可能多的m/z 特征信息来提高信噪比。此外,峰值拾取对于分子生物标志物的识别、量化和发现至关重要[7]。尽管现有的峰值拾取算法已较为成熟,但基线去除、峰宽、信噪比和平滑等操作会引入一定程度的主观性,从而影响生成的峰列表。预处理参数的选择很大程度上依赖于使用者的专业知识,这将导致生物标志物识别的显著差异。
进行峰值拾取后,原始数据的维度降低,但MSI 数据仍具高维复杂性。MSI数据中的一张二维图像通常由数千个高维像素组成,每个像素都有数百个峰值。常用的降维算法是将高维点投影到较小的子空间中,来实现潜在变量的捕获和可视化,从而通过潜在变量揭示分子模式,反映可能具有生物学相关性的相似光谱簇[8]。主成分分析(Principal Component Analysis,PCA)和非负 矩阵分 解(Non-Negative Matrix Factorization,NNMF)的线性降维法已广泛用于MSI数据分析[9-10]。线性降维法的主要限制在于算法的线性约束,不能捕获光谱结构中的非线性特征,进而影响潜在变量的准确识别。与此同时,t 分布随机邻嵌入(t-distributed Stochastic Neighbor Embedding,t-SNE)的非线性降维法近年多被用于组学数据分析[11]。然而,尽管许多研究者在提高t-SNE 计算和内存可扩展性方面取得了进展,但它仍然需要将全部数据加载到RAM 中,这限制了高分辨率MSI 数据集的应用。有研究者对2D MSI 数据集进行峰值拾取后,使用神经网络的自编码器进行降维。与PCA 和NNMF 方法相比,该方法实现了MSI 数据非线性流形的捕获,但是分析速度有限。最近Walid 等[12]开发了一种用于MSI 数据分析和峰值学习的深度学习工具——msiPL,该方法直接将原始MSI 数据输入到模型,且无需进行处理。
本研究在借鉴国内外研究的基础上,以两种不同组织、不同质谱仪器和不同分辨率的MSI 数据为例,基于变分自编码器神经网络的深度学习模型学习并可视化MSI数据底层谱结构的非线性特征;同时通过对复杂分子模式的无监督分析及特定的m/z 值的学习,揭示了小鼠肾脏组织解剖学的生物学相关簇(内髓部、外皮层、肾盂和内皮层小管)和小鼠胃癌模型中的肿瘤异质性,并识别了潜在的特定m/z 峰。通过使用均方误差(MSE)定量评估来评估VAE 模型的学习质量,即评估模型的重建数据与原始数据之间的重建损失。最后,以MSE 为评估指标将msiPL 方法与其他几种专为MSI 开发的高效降维方法(PCA、MemoryEfficient PCA 和DWT+PCA)进行了比较。
1 实验数据
实验中小鼠肾组织数据集来自METASPACE 中的Mouse Kidney 数据集,该数据集由MALDI+FT-ICR 仪器在正离子模式下获得,其中m/z 在100~1998.07 的范围内。采集空间分辨率设置为50μm,包含了21535 个像素,每个像素含5397个m/z。
实验中的方法验证数据集来源于公开数据集[13]。rapifleX MALDI-TOF 仪器,横向分辨率50μm。在采集MALDI-TOF 数据之后,使用7T solariX XR MALDI FT-ICR 分析进行质谱成像的组织切片。MALDI 成像实验是在正离子模式下,通过将m/z 设置在100~3000 范围内,光栅宽度为20μm和15 次激光射击完成的。选择十一肽物质P(RPKPQQFFGLM;[M+H]+,m/z 1347.7354)作为示踪蛋白酶底物,平均强度为23000。原始数据集使用m/z 780.551([PC(34:2)+Na]+)进行内部校准。该数据集包含以下两个数据集:①小鼠胃癌模型MSI 数据集1:没有喷涂蛋白酶底物示踪剂物质P,原始数据包含11006个像素,每个像素是一个高维数据点,每个像素点有4944 个m/z值,即4944 维;②小鼠胃癌模型MSI 数据集2:喷涂蛋白酶底物示踪剂物质P,原始数据包含11823 个像素,每个像素含5138 个m/z值,即5138维。
2 实验方法
2.1 数据预处理
使用R 语言的Cardinal 包对MSI 数据进行总离子流(Total Ion Current,TIC)归一化。在输入msiPL 模型之前要将imzML 标准格式的MSI 数据通过Python 的h5py 包转换为hdf5 格式,并保证输入数据在[0,1]区间内。MSI 数据有不同的归一化策略,最终使用者可以根据实验目的选择最好的MSI 归一化策略,但msiPL 模型的输入数据均需限制在[0,1]内。因为模型的输出层来源于一个sigmoid 激活函数,因此它产生的值在[0,1]之间;同时,输入和输出层的动态范围的一致性对于优化VAE 网络损失函数和最小化重建误差至关重要。
2.2 msiPL模型框架
msiPL 模型基于变分自编码器(Variational AutoEncoder,VAE)结构建立有效的无监督学习、非线性降维和随机变分推理。VAE 网络[14]可同时优化用于变分推理的概率编码器(Encoder)和用于无监督学习的概率解码器(Decoder),如图1所示(彩图扫OSID 码可见,下同)。
编码特征表示在低维空间中学习到的非线性流形,并从原始高维空间捕获分子的空间模式。这些空间模式是基于较小的m/z特征子集形成的,因此识别具有生物学相关性的潜在m/z特征分子至关重要。
基于高斯混合[15](Gaussian Mixture Model,GMM)的编码特征聚类方法计算速度快,并且在识别生物学相关的空间簇(肿瘤和非肿瘤簇)方面非常有效。k是可调参数,可以手动或自动设置。基于信息论的优化过程可用于模型自动选择,但通过手动选择最佳模型可避免遗漏或高估的聚类平衡。由于BIC score 的分布[16]在可搜索范围内逐渐减小,本文将Kneedle 算法(Python,Kneed 包)应用于BIC score 以检测临界点选择最佳模型的最大曲率,即可得到推荐聚类数。
2.3 实现细节
模型输入MSI 数据的高维度特征表示,输出MSI 数据降维之后的特征表示,模型框架如图1 所示。MSI 数据的表示为X={X1,X2,X3,…,XN},其中N为样本数。VAE 网络由输入层、三个隐藏层(h1、h2和h3)和输出层构成。输入层和输出层的神经元个数为m/zbin 的个数;h1、h2和h3的神经元个数分别为512、5、512,其中h2的输出是MSI 数据的5 维空间中压缩的编码特征表示。无监督学习主要通过优化由最大化生成原始数据的概率值和最小化真实和估计后验分布的Kullback-Leibler 散度,KL 散度和VAE loss 函数来最小化原始数据和重构数据之间的重构损失。利用均方误差(Mean Squared Error,MSE)来衡量原始数据和预测数据之间的重构误差。学习率为0.001 的Adam 随机梯度优化器被用于epoch 为100,batchsize 为64的训练网络。VAE 网络基于Keras 的开源深度学习库构建,并在Tensorflow 上运行。
3 实验结果与分析
3.1 小鼠肾脏组织MSI数据集实验
使用msiPL 对来自小鼠肾组织样本的超高光谱分辨率单个MALDI+FT-ICR MSI 数据进行分析。原始MSI数据包含21535 个像素,每个像素是一个高维数据点,每个像素点 的m/z 在100~2000 范围内,包 含l5397 个m/z值,即15397 维。使用msiPL 模型在该数据集上进行降维和可视化实验,神经网络以迭代方式执行无监督学习,以最小化重建损失。如图2(a)所示,优化器在PC 工作站(Intel(R)Core(TM)i7-1065G7 CPU @1.30 GHz 1.50 GHz、16GB RAM、64 位Windows)上经过不到100 次迭代后收敛,总运行时间约为2 h。模型在原始数据和预测数据中的MSE 为3.49×10-4。图2(b)给出了原始数据和预测数据的平均光谱分布,它们的叠加反映了模型的高质量估计。
5 维编码特征表示学习到的非线性特征,可视化并捕获了15397 维的原始高维MSI 数据的分子模式。这些编码特征可用于预测原始数据。MSI 数据的5 维空间中压缩的编码特征表示可视化结果如图3所示。
将学习到的低维嵌入特征使用GMM 模型进行聚类。图4(a)是BIC score 结果图,推荐k为10,也可参考病理学注释结果手动选择设置k的值,研究根据肾组织的生物解剖学分割区域将k为12。BIC score 的范围逐渐减小,然后用Kneedle 算法检测BIC score 的最大曲率临界点。使用GMM(k=12)对整个数据集的编码特征进行聚类识别,每个簇代表一个与小鼠肾脏解剖结构共定位的分子模式,聚类图像如图4(b)所示。聚类图像揭示了不同分子在组织中的空间分布,其聚类结果与参考文献[17]的结果具有一致性。
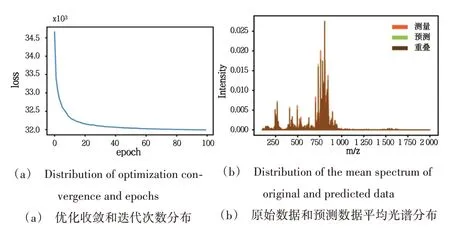
Fig.2 Experimental results of msiPL model on MALDI+FT-ICR MSI data图2 msiPL模型msiPL在MALDI+FT-ICR MSI数据上的实验结果
Fig.3 5-dimensional encoded features visualization results图3 5维编码特征可视化结果
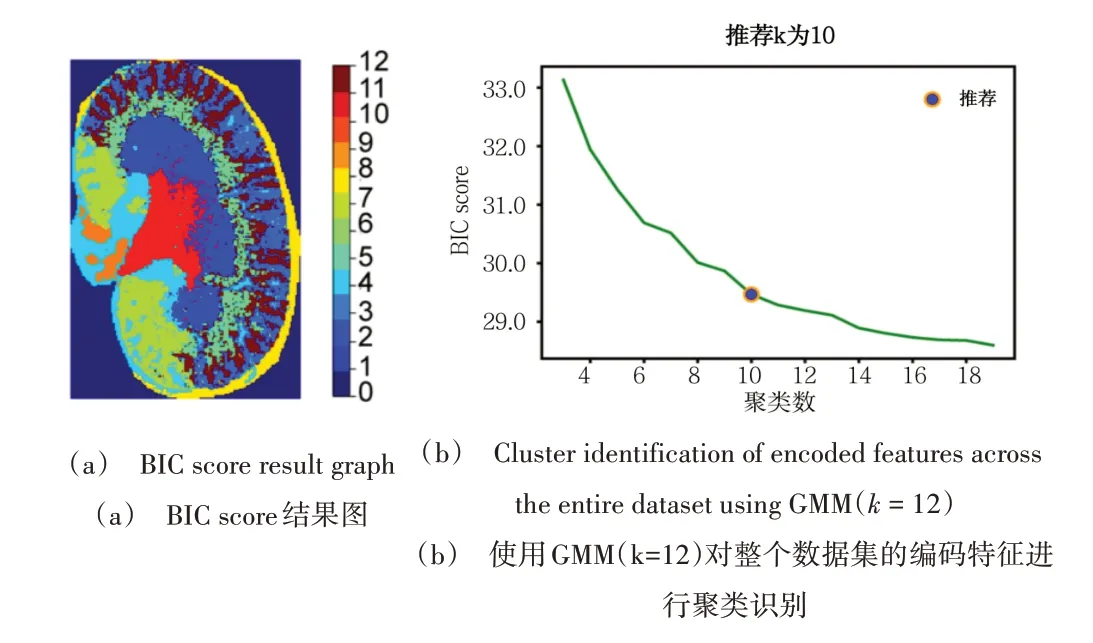
Fig.4 GMM clustering results图4 GMM聚类结果
图5 对每个簇皮尔逊系数最高的m/z离子的空间分布进行了可视化。这些离子与对应簇共定位,如簇1(内髓部)、簇2(外皮层)、簇7(肾盂)、簇9(内皮层小管)。
为了更直观地体现该数据的重建质量,图6 可视化了5 种特定m/z分子的测量数据和重建数据的空间分布。表1为这5种特定分子在METASPACE 平台的注释结果。
将编码的特征链接到原始m/z 变量。拉格朗日乘子β为一个超参数,当β 为1的时候,它即为标准的VAE。一个较高的β 值会降低前变量空间表示信息的丰富度,同时增加模型的解纠缠能力。因此,β 可以作为表示能力和解纠缠能力之间的平衡因子。超参数β 设置在[1,2.5]范围内较好。简化的峰值列表能够揭示潜在空间中捕获的分子模式的主要决定因素。当β 为1 时,学习到439 个m/z;β 为1.5,286个m/z;β为2196个m/z;β为2.5,144个m/z。
3.2 小鼠胃癌模型MSI数据集实验
Katrin 等[13]基于MALDI-MSI 技术探究了小鼠胃癌组织的蛋白酶活性,通过蛋白酶底物示踪剂物质P 的衰减评估内源性蛋白酶活性的空间分布,并提供了小鼠胃癌模型MSI数据集。①小鼠胃癌模型MSI数据集1:没有喷涂蛋白酶底物示踪剂物质P,原始数据包含11006个像素,每个像素含4944 个m/z 值,即4944 维;②小鼠胃癌模型MSI 数据集2:喷涂蛋白酶底物示踪剂物质P,原始数据包含11823个像素,每个像素含5138 个m/z 值,即5138 维。使用msiPL 模型对这两个数据集进行降维和可视化实验。

Fig.5 Visualize the spatial distribution of the m/z ions with the highest Pearson coefficients per cluster图5 可视化每个簇皮尔逊系数最高的m/z离子的空间分布

Fig.6 Spatial distribution of measured and reconstructed data for five specific m/z molecules图6 5种特定m/z分子的测量数据和重建数据的空间分布
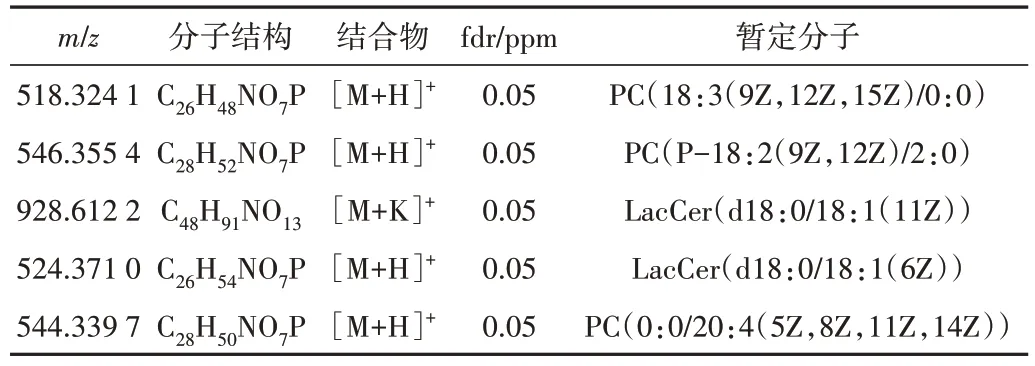
Table 1 Annotation results of five molecules表1 5种分子的注释结果
神经网络以迭代方式执行无监督学习,以最小化损失函数。如图7 所示,优化器在PC 工作站上经过不到100 次迭代后收敛,总运行时间约为40 min。

Fig.7 Optimization convergence and distribution of epochs图7 优化收敛和迭代次数(epoch)的分布
数据集1 和数据集2 的原始数据和预测数据的TIC 归一化平均光谱分布如图8(a)、图8(b)所示。原始数据和预测数据之间的MSE 分别为1.94×10-4、0.70×10-4,重叠的部分反映了高质量估计。

Fig.8 Visualize the distribution of the mean spectrum of the raw and predicted data图8 可视化原始数据和预测数据平均光谱的分布
使用msiPL 模型对数据集2 进行可视化分析。将学习到的低维嵌入特征使用GMM 模型进行聚类,聚类结果如图9(b)所示。将基于GMM 模型的聚类分割结果与H&E注释结果(肿瘤、黏膜、肿瘤—黏膜过渡区和黏膜肌层)进行对比分析,发现msiPL 模型聚类自动分割识别肿瘤和非肿瘤区,H&E 注释结果如图9(a)所示。同时基于GMM 模型的聚类分割结果与基于R 语言的Cardinal 包的空间感知收缩质心聚类分割结果具有一致性,空间感知收缩质心聚类分割结果如图9(c)所示。
重点分析这5 种特定离子:肿瘤标志物脂质离子m/z 798.5(PC(34:1)+K+)、m/z 1347.7(物质P)和肽裂解产物(m/z 1104.6、900.5、753.4),它们的离子图像可视化如图10 所示,同时通过可视化结果评估原始数据与预测数据的重建损失。结合GMM 模型的聚类分割结果和H&E 染色结果分析,发现m/z 为798.54—(PC(34:1)+K+在胃肿瘤区富集,即胃肿瘤组织比非肿瘤组织显示出更显著的蛋白酶活性。与周围黏膜和黏膜肌层相比,肿瘤组织中的P 物质离子强度显着且选择性降低(同时裂解产物m/z 1104.6 和753.4 增加)。相反,肽RPKPQQF(m/z 900.5)在肿瘤中耗尽但在粘膜中富集。值得注意的是,m/z 753.4 的最高浓度区域代表组织外的肽洗脱,此结果体现了抑制剂混合物对P物质裂解和截短肽产生的影响。
3.3 讨论
使用MSE 定量评估来评价VAE 模型的学习质量,即评估模型的重建数据与原始数据之间的重建损失。MSE是一个既定的定量机器学习评估标准,既不主观也不带偏见,因为它是严格的数据驱动的。将msiPL 方法与其他几种专为MSI 开发的高效降维方法(PCA、MemoryEfficient PCA 和DWT+PCA)进行比较,MSE 的结果表明msPL 方法可替代这几种方法进行高效降维,如表2所示。
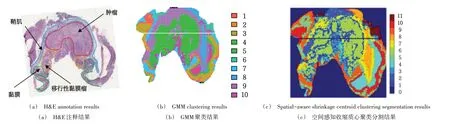
Fig.9 Visualization experimental results图9 可视化实验结果

Fig.10 Spatial distribution of measured and reconstructed data for five specific m/z molecules图10 5种特定m/z离子的测量数据和重建数据的空间分布

Table 2 MSE comparision of MSI data reconstruction using different methods表2 比较使用不同方法 MSI数据重建的均方误差(MSE)
msiPL 模型可实现并行处理和提高内存高效使用。实验中只需将一小部分光谱数据加载到内存中,就可以有效地处理大型和复杂的数据,这体现了对现有计算方法的显著改进。现有计算方法需要将完整数据加载到内存中以计算光谱之间的成对相似性。经典机器学习方法对于高维度原始质谱数据的能力有限,主要是因为这些方法遇到了一个常见的问题,即“维度灾难”,这会降低高维数据的分类精度。相比之下,基于深度学习的方法已经显示出能够避免“维度灾难”并建立相关特征的自学习,从而提高分类准确度。
峰值拾取是一种特征选择,即从原始数据中丢弃大量信息(例如峰形),同时也承担风险丢弃峰选择算法无法识别的峰。这使得后续的质量分析取决于前面的峰值选择或特征选择方法的质量,这可能并不总是理想的。msiPL避免了影响下游分析的预处理步骤。此外,Murta等[18]近期的研究表明峰值拾取参数的选择不仅影响聚类分析,还可能会影响生物学解释。相比之下,msiPL无需优化预处理参数即可实现快速灵敏的MSI数据处理,同时产生可靠的结果。
由于MSI 数据量过大,数据公开集过少,且该研究使用的肾组织样本MSI数据缺乏H&E 染色病理学数据,因此未能将H&E 与MSI离子图像进行共注释分析。
4 结语
经典机器学习方法处理高维度原始质谱数据的能力有限,主要是因为这些方法通常会导致“维度灾难”,降低高维数据的分类精度。相比之下,基于深度学习的方法已经显示出能够避免维度灾难并建立相关特征的自学习,从而提高分类准确度的能力。
本研究在借鉴国内外研究的基础上,以两种不同组织、不同质谱仪器和不同分辨率的MSI 数据为例,使用深度学习数据分析策略实现了从原始高维数据中准确有效地识别和可视化分子模式所需的底层非线性流形,避免了传统的、耗时的预处理计算。同时通过对复杂分子模式的无监督分析及特定的m/z 值的学习,揭示了小鼠肾脏组织解剖学的生物学相关簇(内髓部、外皮层、肾盂和内皮层小管)和小鼠胃癌模型中的肿瘤异质性,并识别了潜在的特定m/z峰。由于MSI 数据量过大,数据公开集过少,且该研究使用的肾组织样本MSI数据缺乏H&E 染色病理学数据,未能将H&E 与MSI 离子图像进行共注释分析。msiPL 在MSI数据集无监督分析和识别生物相关性的空间模式方面的高效处理能力,使msiPL 模型直接从质谱数据中完成对肿瘤类型和等级的分类和预测在未来成为可能。
