黑龙江省和海南省稻瘟病菌中AVR-Pita家族的分布及变异分析
2023-03-07刘瑞赵羽涵顾欣怡王艳霞靳学慧吴伟怀张亚玲
刘瑞,赵羽涵,顾欣怡,王艳霞,靳学慧,吴伟怀,张亚玲
黑龙江省和海南省稻瘟病菌中家族的分布及变异分析
刘瑞1,赵羽涵1,顾欣怡1,王艳霞1,靳学慧1,吴伟怀2,张亚玲1
1黑龙江八一农垦大学农学院/黑龙江省植物抗性研究中心,黑龙江大庆 163319;2中国热带农业科学院环境与植物保护研究所,海口 571101
【目的】通过检测黑龙江省和海南省不同稻瘟病菌()菌株群体中家族的分布情况和变异特征,了解其变异类型的致病表型,为区域内抗病种质的筛选与选育提供参考。【方法】通过参考NCBI中公布的家族序列分别对启动子区和CDS区设计特异性引物共6对,对2020年采自黑龙江省和海南省不同地区的397个稻瘟病菌单孢菌株提取DNA,进行PCR扩增。通过电泳检测结果,分别选取囊括不同地区的代表菌株对扩增片段进行测序。测序结果与NCBI中相应的碱基与氨基酸序列进行比较分析,并利用水稻抗性单基因系,对不同变异类型的稻瘟病菌菌株进行无毒功能验证。【结果】在PCR电泳检测结果中,黑龙江省稻瘟病菌菌株携带所有待测基因,分布广泛且检出频率较高;而海南省稻瘟病菌菌株中仅携带和,并以低频率集中存在。无毒基因组成分析结果显示,黑龙江稻区的稻瘟病菌菌株复杂多样,携带的基因型种类较海南菌株丰富。PCR产物测序检测结果显示,家族以点突变、插入和缺失为主要变异类型划分为19种,且不同稻瘟病菌群体来源的菌株,其变异类型具有特异性。检测出10种变异类型,其中(1—5)为黑龙江省稻瘟病菌菌株独有的变异类型,(6—10)为海南省稻瘟病菌菌株独有。对这10种变异类型经功能验证后发现,无毒功能均已丧失。检测出8种变异类型,其中(1—4)为黑龙江省稻瘟病菌菌株独有,(5—8)为海南省稻瘟病菌菌株独有。经功能验证后发现,无毒功能均已丧失。在黑龙江省仅检测出1种变异类型。【结论】黑龙江和海南稻瘟病菌群体中家族均为突变后的等位基因型存在,经致病性鉴定后,检测出的所有突变类型均不能被相对应抗性基因和Pi-ta所识别,表现为感病。因此,抗性基因和Pi-ta在黑龙江省和海南省对稻瘟病的抗病育种与利用过程中可聚合其他抗性基因应用来保证品种抗病性。同时,不同地理来源的稻瘟病菌菌株群体中家族的分布和变异类型具有特异性。
稻瘟病菌;家族;变异;无毒基因;黑龙江省;海南省
0 引言
【研究意义】由稻瘟病菌(无性态:)侵染引起的稻瘟病是对水稻生产造成巨大威胁的三大病害之一[1-2]。选育和种植抗性品种被证明是控制稻瘟病最经济有效和环境无害的途径。水稻与稻瘟病菌之间符合经典的“基因对基因”系统[3],它解释了病原菌中的无毒基因()与水稻中特定的抗性基因()相对应,以及品种中缺失某抗性基因如何使该品种成为感病品种[4]。但由于稻瘟病菌生理小种的复杂多变及无毒基因的频繁变异,不同稻瘟病菌群体遗传结构发生变化,产生新的无毒基因型,最终导致水稻品种抗性“丧失”[5]。我国地貌纷繁,生态环境多样,致使不同稻瘟病菌菌群中无毒基因结构复杂,遗传变异类型多样[6]。因此,监测不同地域稻瘟病菌群体中无毒基因的分布及变异情况,了解无毒基因变异对病原物无毒功能丧失的机制,对研究不同稻区稻瘟病菌菌群的遗传多样性和区域内指导抗病育种方向,并实现抗病持久化具有重要意义。【前人研究进展】据不完全统计,目前已鉴定的无毒基因有40多个,已被克隆的无毒基因有12个,分别为、[7-16]。Orbach等[7]通过PFLP标记克隆了无毒基因,定位在稻瘟病菌第3号染色体近端粒处,该基因编码一个含223个氨基酸的分泌蛋白,含典型锌金属蛋白酶结构。由于端粒具有不稳定性,常发生点突变、插入和缺失等变异来逃脱的识别,使水稻抗性丧失。Jia等[17]通过蛋白质印迹证明是首个编码蛋白与相应抗病基因产物直接互作的无毒基因。Khang等[18]研究发现,是一个基因家族,包括、和,其中为原来的基因。研究还发现,分别与和具有92%和71%的DNA序列同一性。和具有与相对应的无毒功能,且两基因均存在于种群和种群中,而仅存在于种群中。一些菌株中的和位于端粒附近,两侧是反向重复DNA元件,两基因的频繁缺失和扩增可能是重复DNA元件发生重组所引起的。在水稻稻瘟病抗性基因克隆方面,迄今为止已报道了40个抗瘟基因并完成了分子克隆[19],与Pi-ta均定位于水稻12号染色体的着丝粒区域。目前虽已成功克隆,但与Pi-ta仍然不能在F2代中有效分离,因此猜测两基因可能紧密连锁或等位基因[19]。Bryan等[20]发现,对识别特异性的差异取决于Pi-ta蛋白之间的单个氨基酸差异。研究还表明等位基因也出现在含有Pi-ta的水稻品种中,实际上就是Pi-ta为至少加上一个紧密连锁的其他抗性基因。甘玉姿等[21]在菲律宾稻瘟病菌中发现一个新的等位基因,命名为,研究还发现和不具有无毒功能。余欢等[22]在国内草类寄主梨孢菌中发现,具有丰富的变异性,在基因上游存在大量序列缺失现象,基因下游存在缺失、部分缺失和多位点现象。【本研究切入点】水稻在中国种植范围广,南起海南岛,北至黑龙江,分别以种植籼稻和粳稻为主。黑龙江省地处东北早熟单季稻稻作区,属于寒温带与温带大陆性季风气候;海南省地处华南双季稻稻作区,属于热带季风海洋性。受种植区内生态、地势等因素影响,区域内稻瘟病菌的无毒基因可能会存在差异。笔者实验室已报道了黑龙江省和海南省基因家族在稻瘟病菌中的分布及变异情况[23],但目前针对南北不同地域差异的稻瘟病菌群体中家族的分布变异情况的研究鲜有报道。因此,为探究南北不同稻瘟病菌菌群中家族的分布及变异机制,对采自黑龙江省和海南省不同水稻产区的稻瘟病菌进行分析。【拟解决的关键问题】通过对黑龙江省和海南省稻瘟病菌中家族进行PCR检测、基因序列测序以及对基因的不同变异类型进行功能验证,明确这两个稻区内稻瘟病菌家族的分布差异及变异特性,为区域内抗病种质的筛选与选育提供参考。
1 材料与方法
1.1 试验材料
稻瘟病菌供试群体均为2020年于黑龙江省7市14县和海南省5市6县采集的稻瘟病穗颈瘟标样经分离获得的397个单孢菌株,采集地点及编号如表1。
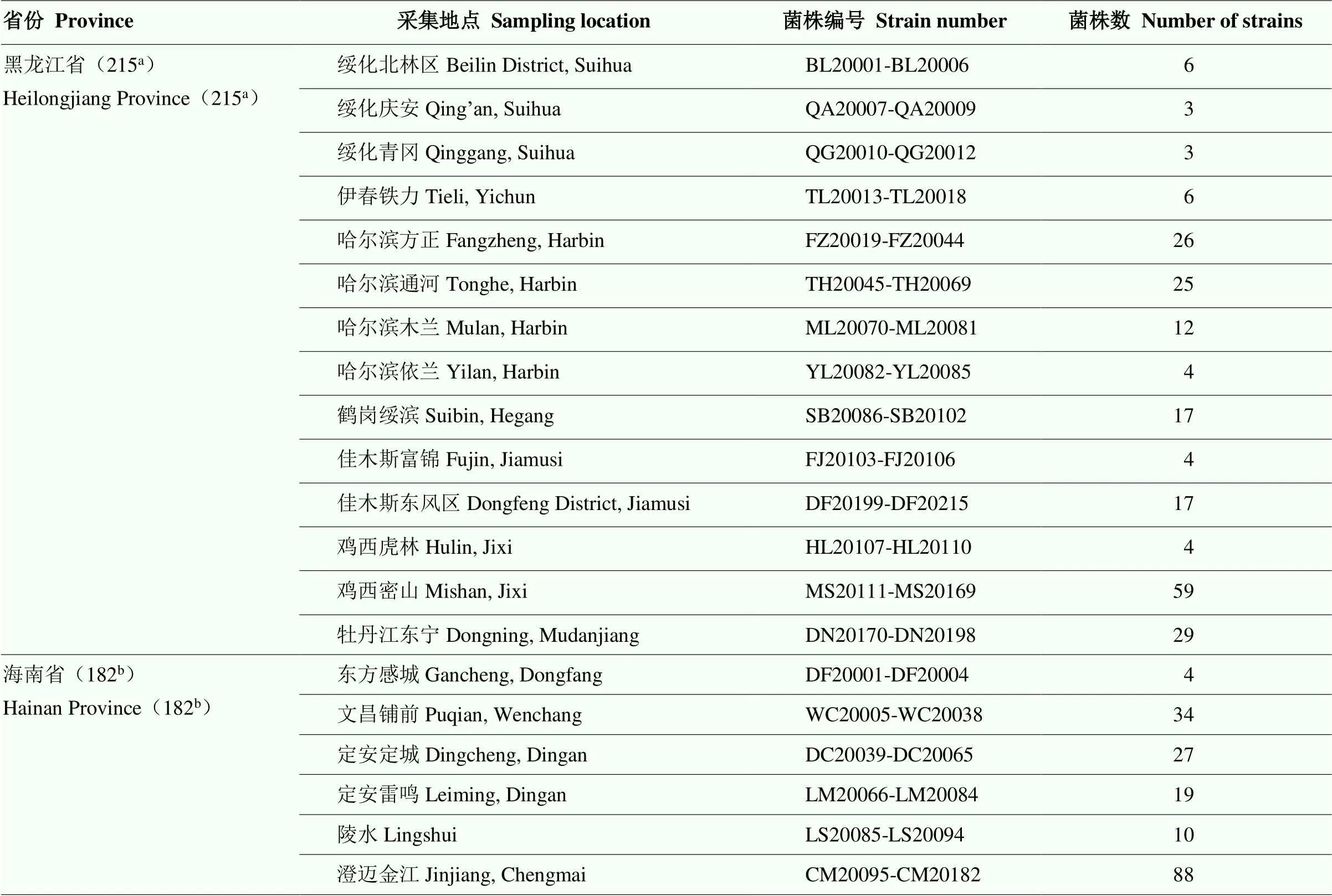
表1 黑龙江省和海南省稻瘟病菌菌株
a:黑龙江省菌株数Number of strains in Heilongjiang Province;b:海南省菌株数Number of strains in Hainan Province
1.2 稻瘟病菌基因组DNA的提取
将供试的稻瘟病菌单孢菌株置于PDA平板培养基上活化处理,取5块菌碟放入灭菌的150 mL液体酵母培养基内,置于立式恒温振荡培养箱摇菌一周,用真空泵抽滤菌丝后分装于离心管中。使用CTAB[24]法提取菌丝DNA。采用核酸蛋白测定仪检测提取的DNA浓度和纯度,以确保其质量,并将DNA原液用TE稀释成50 ng·µL-1备用,置于-20℃保存。
1.3 引物设计
于NCBI(https://www.ncbi.nlm.nih.gov)上查找稻瘟病菌家族的基因序列,利用Primer premier 5设计3对特异性引物,启动子区序列引物参考Khang等[18],编码序列(CDS)区序列引物参考孟峰等[25],编码序列(CDS)区序列引物参考Shi等[26]。6对引物均委托生工生物工程(上海)股份有限公司合成(表2)。

表2 用于扩增无毒基因的引物
1.4 PCR扩增及电泳检测
PCR反应体系(20 μL):dNTPs 1.6 μL,10×Buffer 2 μL,正、反引物各0.3 μL,模板DNA 1 μL,rTaq酶0.1 μL,ddH2O补足至20 μL。
PCR扩增程序:95℃预变性4 min,94℃变性45 s,50—60℃退火45 s,72℃延伸30—90 s,35个循环,72℃延伸5 min,4℃保存,待检测。
电泳检测:PCR产物在电泳液为0.5×TBE和琼脂糖配置好的1%琼脂糖凝胶中电泳20 min,150 V。在电泳成像仪下利用Image Lab系统中的条带检测功能读取并记录电泳条带,根据电泳图谱,运用二进制方法,将有条带的记为“1”,没有带的记为“0”。将数据制成Microsoft Excel文件,并对文件中的数据进行分析。根据检测结果计算各稻区稻瘟病菌中无毒基因家族的检出频率。
1.5 无毒基因的基因序列分析
从无毒基因引物扩增的PCR产物中挑选不同带型不同水稻产区的部分菌株,送至生工生物工程(上海)股份有限公司测序,测序结果采用DNAMAN软件对碱基及核苷酸的序列进行比对分析,确定变异类型。
1.6 稻瘟病菌菌株致病性测定
水稻品种:含有、Pi-ta抗性基因的水稻单基因系IRBLta-K1和IRBLta2-Pi,为国际水稻研究所选育;感病对照品种:丽江新团黑谷(LTH)。
菌株活化及产孢培养:在PDA平板培养基上将供试的稻瘟病菌单孢菌株活化处理,5 d后接种到米糠培养基上进行产孢培养,待菌丝长满培养基表面时,用涂布棒和无菌水刮洗掉气生菌丝,置于恒温恒湿环境中,用蓝紫光灯照射3 d,进行产孢培养。
水稻种植:挑选籽粒饱满的水稻种子用1.25‰的多菌灵溶液室温下浸泡24 h,用清水冲洗干净后置于滤纸保湿的培养皿内,于30℃的恒温培养箱催芽。将发芽后的种子播种于固定在塑料水培盒(尺寸:225 mm×155 mm×55 mm)内的定植篮(上端内径34 mm,下端内径28 mm,高45 mm)中,每个定植篮内播种10粒,自来水培养3 d后,营养液培养,营养液采用经典的霍格兰(Hoagland)配方,每4 d换一次营养液,待水稻3叶1心时进行人工喷雾接种。
接种调查:待米糠培养基表面产生灰色霉层后,每个培养皿用5 mL无菌水洗下孢子,用双层纱布过滤,制成孢子悬浮液,在40倍显微镜下每视野孢子量大于40个,加入1%的吐温20,摇匀后均匀地喷洒在秧苗上,将其移入25℃的遮光保湿棚内培养24 h后返回室温自然培养,每组处理重复2次,接种5 d后调查植株发病情况,调查标准参考靳学慧[27]。
2 结果
2.1 AVR-Pita基因家族在黑龙江省和海南省稻瘟病菌中的分布
利用基因家族启动子区和CDS区序列引物对397个稻瘟病菌菌株DNA进行PCR检测(图1),电泳结果显示,基因家族共出现3种带型不同的条带:有带、无带和低带,用“+”“-”和“1”表示。在黑龙江省186个菌株中扩增出特异性片段,检出频率为86.51%;在海南省90个菌株中均扩增出特异性片段,检出频率为49.45%,其中东方感城、澄迈金江检出频率为0。在黑龙江省178个菌株中扩增出特异性片段,检出频率为82.79%;在海南省89个菌株中均扩增出特异性片段,检出频率为48.90%,其中东方感城、澄迈金江检出频率为0。仅在黑龙江省186个菌株中扩增出特异性片段,检出频率为86.51%;而海南省菌株中均未增出特异性片段,检出频率为0。
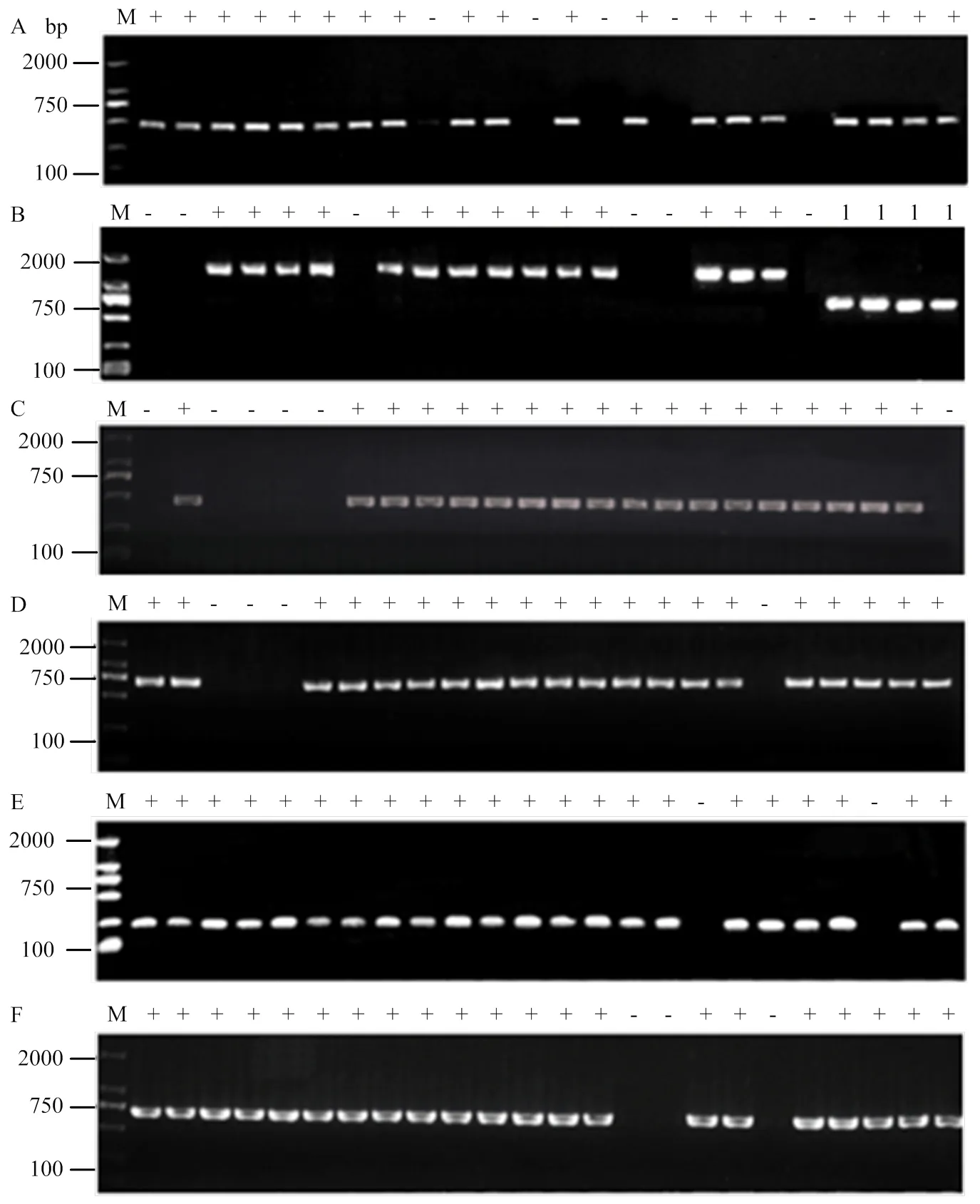
A:AVR-Pita1启动子区的部分扩增结果Partial amplification results of AVR-Pita1 promoter region;B:AVR-Pita1 CDS区的部分扩增结果Partial amplification of AVR-Pita1 CDS region;C:AVR-Pita2启动子区的部分扩增结果Partial amplification results of AVR-Pita2 promoter region;D:AVR-Pita2 CDS区的部分扩增结果Partial amplification of AVR-Pita2 CDS region;E:AVR-Pita3启动子区的部分扩增结果Partial amplification results of AVR-Pita3 promoter region;F:AVR-Pita3 CDS区的部分扩增结果Partial amplification of AVR-Pita3 CDS region;M:DNA marker DL2000;1:低带Low band;+:存在Existence;-:缺失Missing
2.2 AVR-Pita基因家族在黑龙江省和海南省稻瘟病菌中的基因型
基因家族广泛分布于所有被调查的稻瘟病菌菌株中,单个基因的存在和不同组合的分析如表3所示。397个菌株中共检测到7种不同基因型,分别为、、+、+、+和++。黑龙江省菌株含有全部组合类型,其中携带家族的3个无毒基因的菌株数量最多,为155株,占72.09%且均以组合形式存在。而海南菌株仅有2种类型,和+,其中不携带待测家族基因的菌株数量最多,为92株,占50.55%;携带+基因型的菌株数量为90株,占49.45%,单个家族基因不单独存在。由此可见,黑龙江菌株携带的基因型种类较海南菌株丰富。
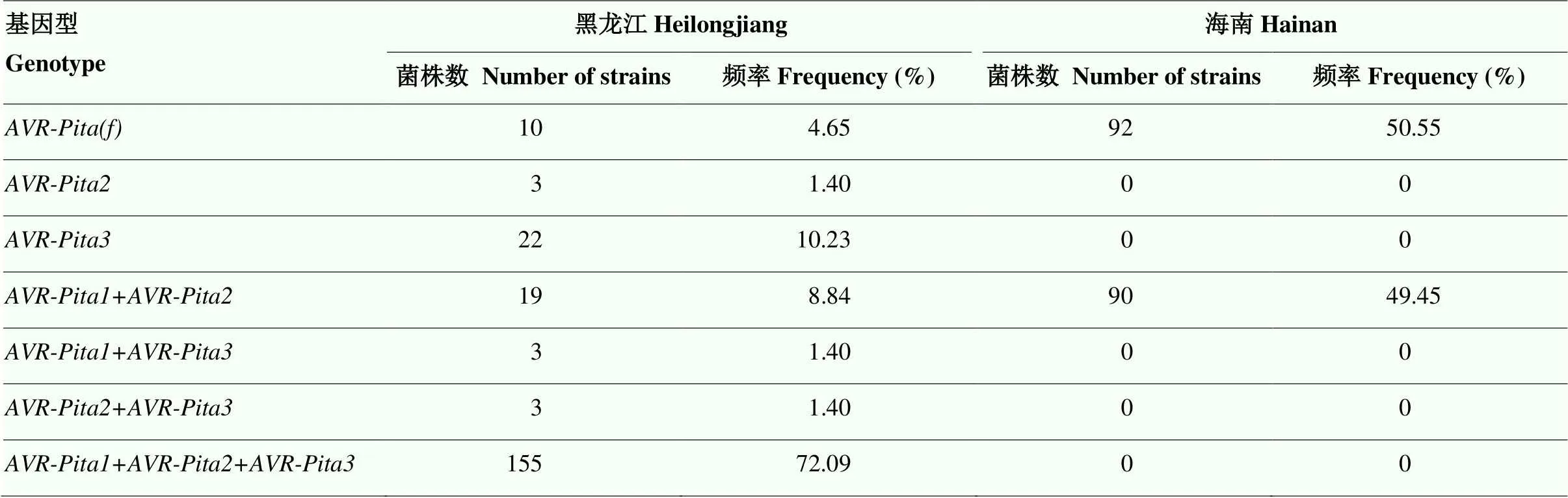
表3 参试菌株携带无毒基因型的检测结果
:不含待测基因的基因型,其中,f表示不存在Genotypes that do not contain the gene to be tested, where, f stands for free
2.3 AVR-Pita基因家族的序列及致病表型分析
2.3.1的序列及致病表型分析 分别于两省各选取20个囊括所有地区,并在启动子区和CDS区均扩增出特异性片段菌株的PCR产物送去测序,将测序结果与的标准序列进行比对。结果显示,用于测序的40个菌株表现出10种序列类型(表4),第1类同余欢等[22]发现的变异类型一致(),与标准序列比对,在启动子区-1 004 bp至CDS区+246 bp处存在1 250 bp基因序列的缺失;其余9类的差异主要体现在碱基的替换与插入,即-413(A/G)、-376(G/A)、-246(T/C)、-116(C/A)、-52(A/C)、18-20GTT(Insert)、207(C/T)、245(G/A)、247(A/G)、263(A/G)、312(T/G)、407(A/G)、520(A/G)、575(G/A)/(T/A)、580(C/T)和583(C/G),其中575位点处A碱基突变了2种类型,将这9种变异类型分别命名为、、、、、、、和。
的9类碱基序列中,-(2,3)、-(6,7,8)和-(9,10)变异类型在CDS区变异位点一致,因此从中选取、和的氨基酸序列参与序列比对。将5类碱基序列所翻译的氨基酸序列与标准氨基酸序列进行比对(图2),结果显示,207(C/T)处为同义突变,其余碱基突变均使氨基酸发生错义。在6(L/Insert)、82(S/N)、83(N/D)、88(K/R)、104(N/K)、174(I/V)和192(C/Y)处发生氨基酸错义翻译;在6(L/Insert)、82(S/N)、83(N/D)、88(K/R)、104(N/K)、136(E/G)、174(I/V)和192(C/Y)处发生氨基酸错义翻译;在6(L/Insert)、82(S/N)、83(N/D)、88(K/R)、104(N/K)、136(E/G)和174(I/V)处发生氨基酸错义翻译;在类型的基础上增加一处,即194(R/W);在6(L/Insert)、192(F/Y)和195(H/D)处发生氨基酸错义翻译。
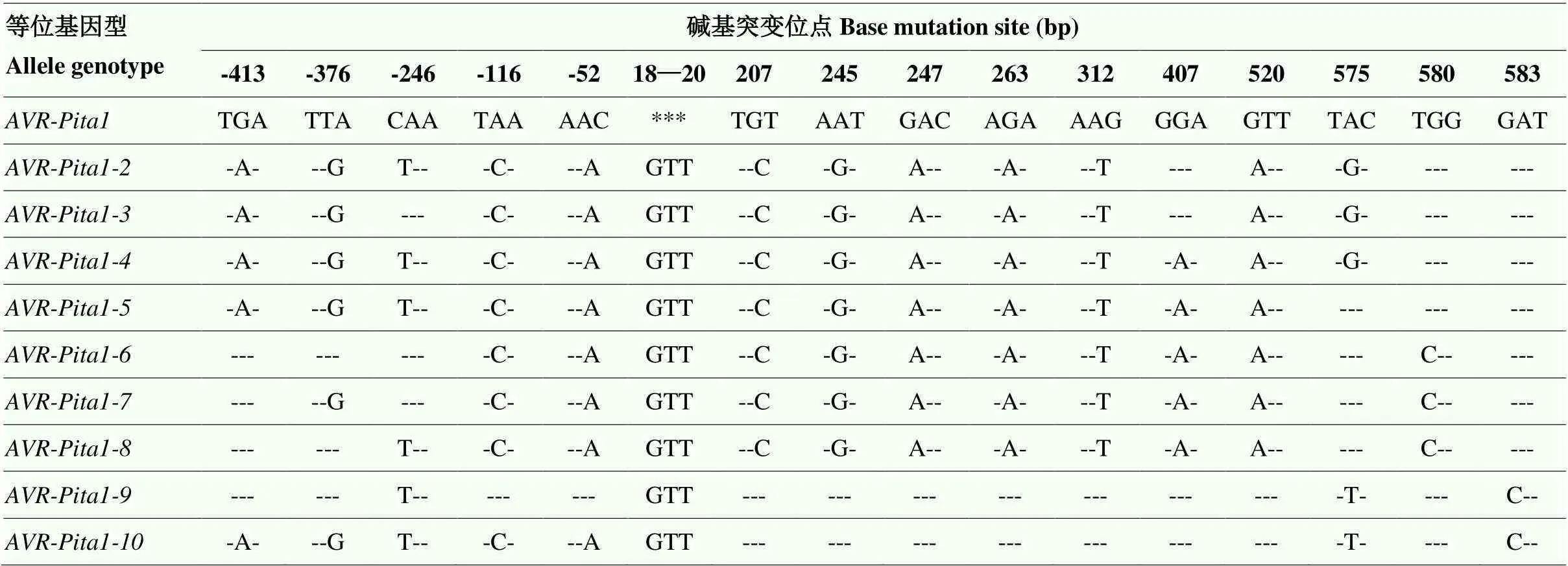
表4 部分菌株AVR-Pita1碱基序列比对结果
-代表在该点的碱基与标准序列相同- represents that the base at this position is the same as the standard sequence

图2 AVR-Pita1氨基酸序列比对分析
对无毒基因型的10种突变类型的菌株接种水稻单基因系K1()进行功能验证,以丽江新团黑谷(LTH)为对照。结果显示(图3),-(1—10)均不能被识别而表现为有毒性(V),无毒功能丧失。
2.3.2的序列及致病表型分析 分别于两省各选取20个囊括所有地区,并在启动子区和CDS区均扩增出特异性片段菌株的PCR产物送去测序,将测序结果与的标准序列进行比对。结果显示,用于测序的40个菌株表现出8种序列类型(图4),均与标准序列有多处差异,主要体现在单碱基的替换、插入与缺失,其中,-115、506和575位点处单碱基各突变了2种类型,即-115(C/T)/(A/T)、506(G/A)/(T/A)和575(G/A)/(T/A)。第1类、第2类和第4类各与标准序列相比均有89处差异,将这3类分别命名为、和;第3类和第7类各与标准序列相比均有88处差异,将这2类分别命名为和;第5类、第6类和第8类各与标准序列相比分别有86、85和87处差异,将这3类分别命名为、和。

图3 AVR-Pita1变异菌株的致病型

图4 部分菌株AVR-Pita2碱基序列比对结果
将的8类碱基序列所翻译的氨基酸序列与标准氨基酸序列进行比对(图5),结果显示,8种变异类型均与标准氨基酸序列有多处差异,均为碱基突变使氨基酸发生错义。其中,88、169和192位点处各错义突变了2种类型,即88(K/E)/(R/E)、169(G/Q)/(V/Q)和192(C/Y)/(F/Y)。(1,2,4,7)均与标准序列比对有33处差异;(3,8)均与标准序列比对有32处差异;和与标准序列比对分别有31和30处差异。所有变异位点处均发生氨基酸错义翻译。
对无毒基因型的8种突变类型的菌株接种水稻单基因系PiNo.4(Pi-ta)进行功能验证,以丽江新团黑谷(LTH)为对照。结果显示(图6),-(1—8)均不能被Pi-ta识别而表现为有毒(V),无毒功能丧失。
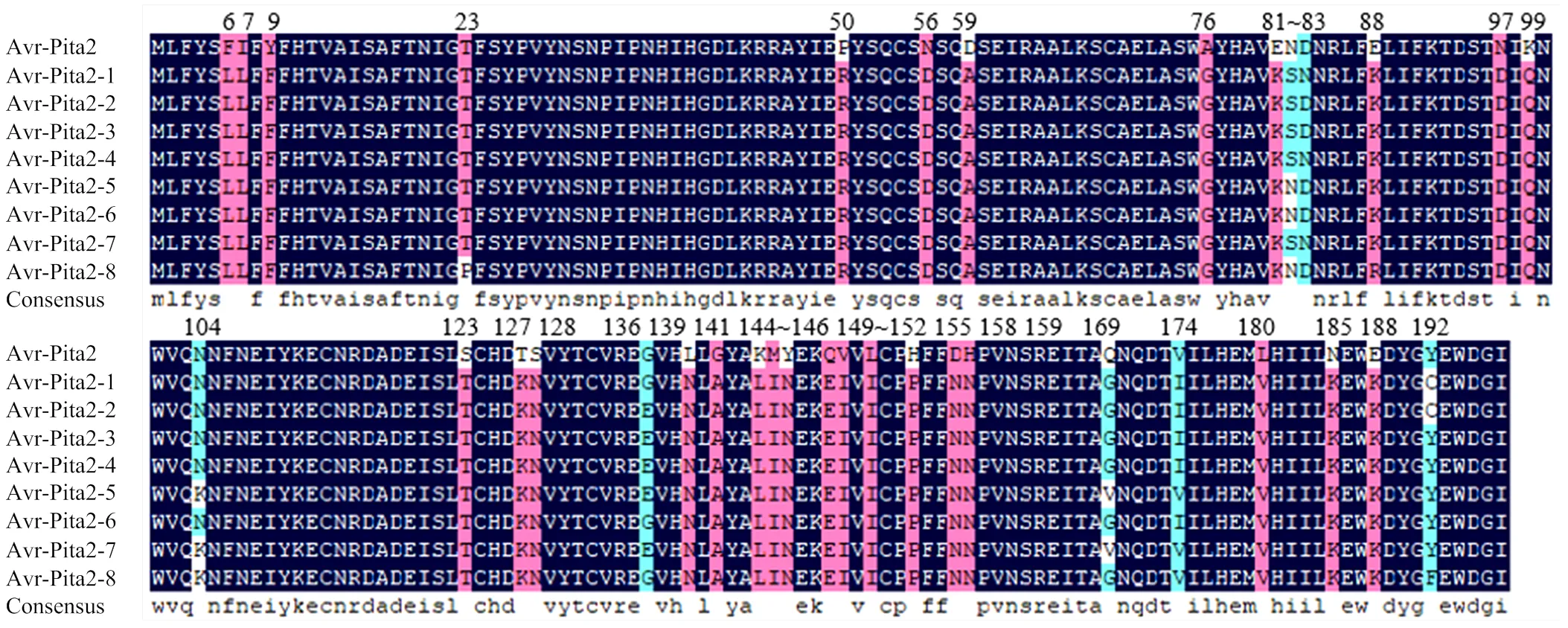
图5 AVR-Pita2氨基酸序列比对分析

图6 AVR-Pita2变异菌株的致病型
2.3.3序列分析 因海南菌株中未扩增出目的片段,故于黑龙江菌株中选取20个囊括所有地区,并在启动子区和CDS区均扩增出特异性片段菌株的PCR产物送去测序,将测序结果与的标准序列进行比对。结果显示,用于测序的20个菌株仅表现出1种序列类型(表5),与标准序列比对有多处差异,主要体现在单碱基的替换,即55(G/A)、103(C/T)、122(T/C)、135(A/G)、148(C/A)、183(C/A)、267(A/C)、307(A/G)、315(T/C)、319—321(CGT/AAG)、333(C/A)、479(G/A)、484(A/G)和508(G/A)。
将碱基序列所翻译的氨基酸序列与标准氨基酸序列进行比对(图7),结果显示,135(A/G)和315(T/C)处为同义突变,其余碱基突变均使氨基酸发生错义,即19(V/I)、35(P/S)、41(V/A)、50(Q/K)、61(D/E)、89(L/F)、103(K/E)、107(R/K)、111(D/E)、160(R/Q)、162(K/E)和170(A/T)。
2.3.4基因家族变异类型占比 在测序结果中,基因家族序列类型可划分为19种变异类型,其在黑龙江省和海南省占比如图8所示。-(1—5)仅在黑龙江省存在,占比较均匀,分别为30.00%、25.00%、5.00%、25.00%和15.00%;反之-(6—10)仅在海南省存在,其中-占比最大,为50.00%,其余基因型占比分别为15.00%、10.00%、5.00%和20.00%。-(1—4)仅在黑龙江省存在,其中占比最大,为70.00%,其余基因型占比分别为10.00%、5.00%和15.00%;反之-(5—8)仅在海南省存在,其中占比最大,为55.00%,其余基因型占比分别为20.00%、15.00%和10.00%。仅在黑龙江菌株中检测到,且所有测序菌株均有同一种变异类型。以上结果说明南北不同稻瘟病菌菌株群体中家族具有显著差异,并均含有其特异性变异类型,即不同地理及品种来源的菌株存在另一地区不存在的变异类型,并且不同菌群来源的菌株具有丰富的遗传多样性。
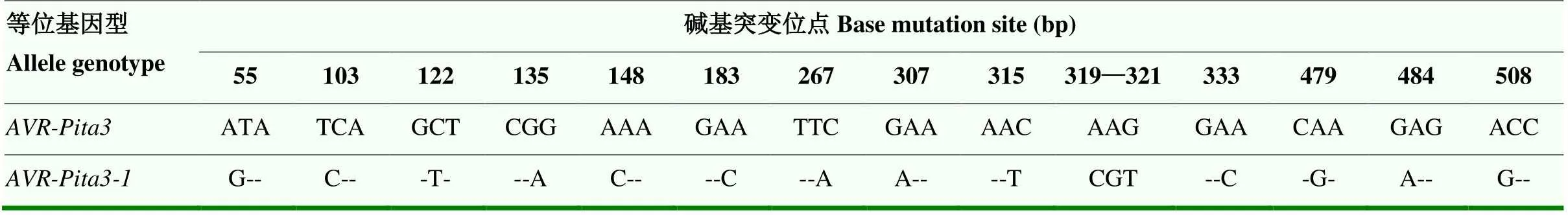
表5 部分菌株AVR-Pita3碱基序列比对结果
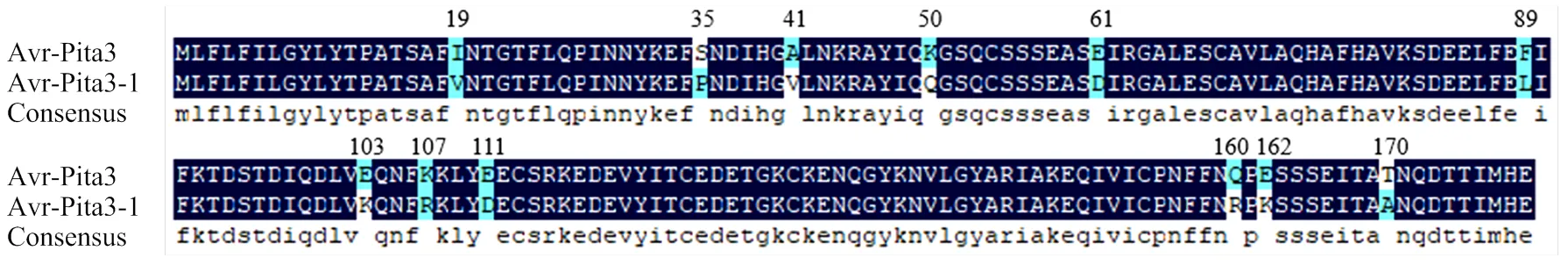
图7 AVR-Pita3氨基酸序列比对分析
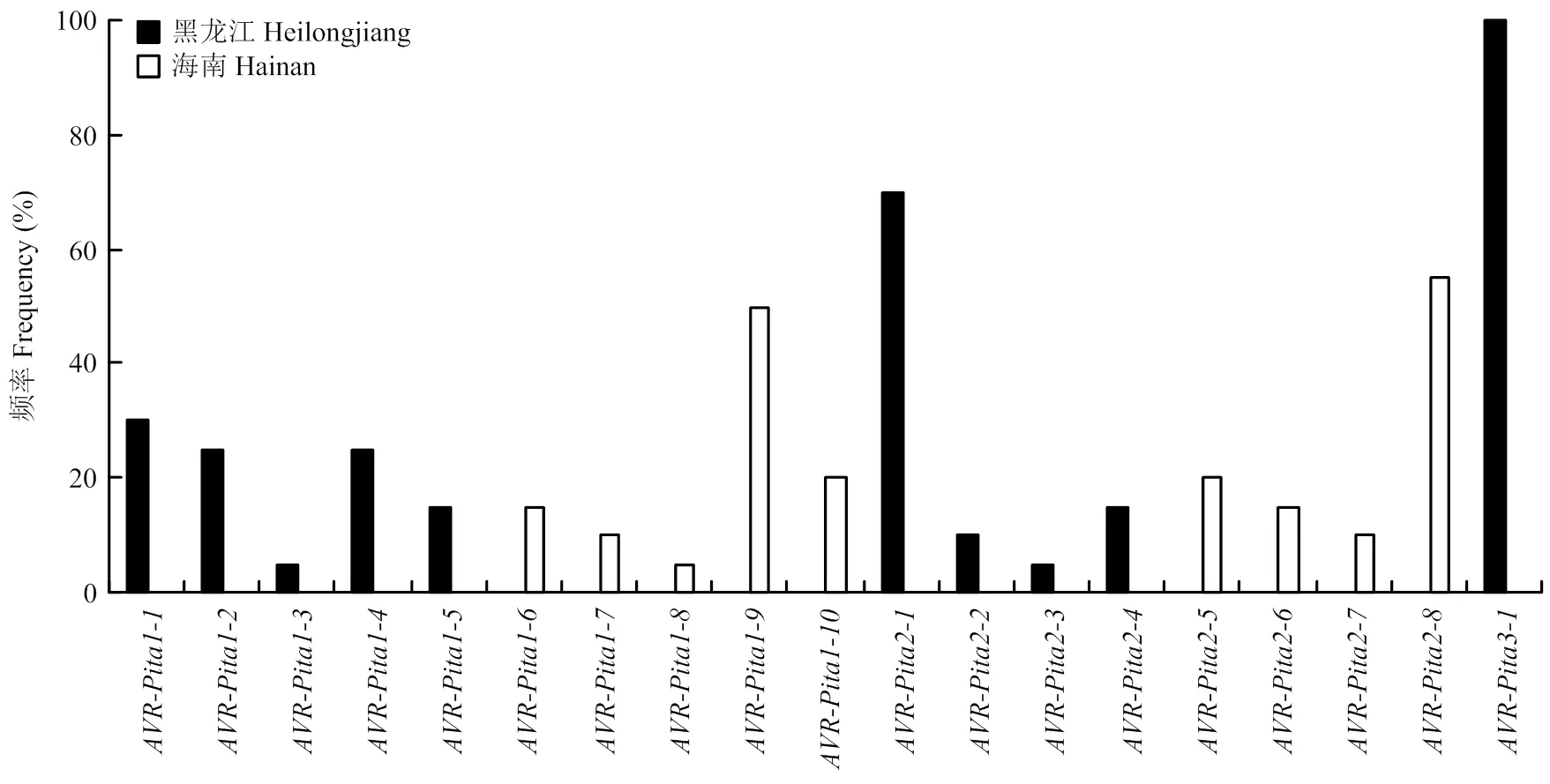
图8 AVR-Pita基因家族变异类型比例分析
3 讨论
3.1 黑龙江省和海南省稻瘟病菌无毒基因型的分布
黑龙江省和海南省分别是中国粳/籼稻的主产稻区,由于我国生态环境的差异,稻瘟病菌极易通过熟悉的抗性条件衍生出不同的致病型,同时,抗病品种的大面积单一化种植也会对稻瘟病菌造成选择压力,促使其发生变异导致品种抗性“丧失”[28],进而使水稻大幅减产甚至绝收。Valent等[29]研究表明,水稻品种的持久抗性与所处稻区的稻瘟病菌无毒基因的变化相关。因此,及时检测不同稻瘟病菌群体中无毒基因的分布和无毒基因型的组成情况,可为不同稻区合理进行抗病基因应用提供科学依据。
本试验中检测到大部分黑龙江省水稻种植区内,稻瘟病菌菌株中携带所有待测基因,且出现频率较高,表明家族在该地区广泛存在;而少数海南省水稻种植区内,稻瘟病菌菌株中携带和,出现频率较低,且所有海南菌株中均不携带,由于本文供试的海南菌株均采自稻瘟病发生较重且具有代表性的地区,因此,推测海南菌株中不存在或含有较少的,表明该家族在海南地区以低频率集中存在并具有地域差异性。这些结果表明,黑龙江和海南省粳/籼稻不同稻瘟病菌群体中所检测的家族分布差异较大,与廖静静等[30]对云南省粳/籼型稻瘟病菌中主要无毒基因分布的研究结果相吻合,表明粳/籼型稻瘟病菌无毒基因的分布是稻瘟病菌对水稻亚种适应性进化的结果。对每个菌株的无毒基因组成进行分析,结果显示,不同稻瘟病菌群体中同一无毒基因型的出现频率存在差异,与房文文等的研究结果一致[31-32]。所有供试菌株可划分为7种无毒基因型,黑龙江菌株含有全部组合类型;而海南菌株仅含有2种类型,说明黑龙江稻区的稻瘟病菌菌株中携带的基因组合种类较为丰富,而海南稻区较为单一。稻瘟病菌群体无毒基因分布形成明显区域特征的原因可能是由于各地区均有其独具的生态条件,长期以来经自然选择的结果,使不同地区稻瘟病菌群体产生了独特的无毒基因结构。
周江鸿等[33]研究表明,我国稻瘟病菌群体对携带抗性基因的品种表现为中等至强毒力水平,因此推测家族存在高变异性,易失去无毒功能。与本研究结果一致,黑龙江和海南稻瘟病菌群体中家族均为突变后的等位基因型存在,经致病型鉴定后,检测出的所有突变类型均不能被相对应抗性基因和Pi-ta所识别,表现为感病。高清等对粳型水稻中抗瘟基因分布和抗病性的研究结果表明,抗性基因在黑龙江省水稻品种中总体应用较广,聚合其他抗性基因的抗病率较高,认为抗性基因在黑龙江省水稻抗病育种中具有较高贡献[34-35]。孟峰等研究表明,黑龙江省各稻区稻瘟病菌中变异类型相对稳定,认为在黑龙江省水稻抗病育种中,需要聚合其他抗性基因才能提高抗病率[25,36],与本文研究结果一致,抗性基因和Pi-ta在黑龙江省水稻的抗病育种与利用过程中需要聚合其他抗性基因应用来保证品种抗病性。董丽英等[37]研究表明,云南省籼稻中和Pi-ta的抗性频率均高于80%,两抗性基因可作为抗原在该地区应用;曹雪琦[38]研究表明,福建省籼稻中Pi-ta的抗性频率低于30%,携带该基因的水稻品种在福建省应慎重推广;周瑚等[39]研究表明,湖南省籼稻中和出现频率低,且和Pi-ta的抗性频率均在40%左右,携带两单基因的水稻品种在湖南省应慎重推广。本研究中,海南省稻瘟病菌菌株中和出现频率低,因此抗性基因和Pi-ta在海南省对稻瘟病的抗病育种应用中同样需要慎重考虑,可聚合其他抗性基因应用来保证品种抗病性。
3.2 黑龙江省和海南省稻瘟病菌无毒基因型的进化
对遗传变异进行评估是了解基因进化和稻瘟病菌与水稻共同进化的主要分子机制。由于家族基因位于不稳定的端粒处,并在稻瘟病菌1、3—7号等染色体上均有发现,导致该基因具有高度的可变性和多态性,进而导致更高水平的遗传多样性。家族的变异可发生在基因的CDS区、5′端及端粒区[40],任何通过点突变、插入与缺失等细微的变异都可以影响整个分泌蛋白的结构,对无毒功能造成影响,最终达到改变其致病性的目的。因此,了解不同菌株群体中无毒基因的变异机制对掌握其不同遗传多样性和对区域内抗性品种的选育是至关重要的。
通过挑选不同群体中家族的PCR产物测序后发现,家族在不同群体中有着丰富的遗传多样性和特异性。Sirisathaworn等[41]报道了基因突变是影响稻瘟病菌群体遗传多样性的唯一因素,随后作者对泰国稻瘟病菌中3个的编码序列进行分析发现,3个均呈现较低水平的遗传变异,即泰国稻瘟病菌具有低水平的遗传多样性。Kasetsomboon等[42]报道了编码区存在15个单倍型,且具有高度的遗传多样性。本研究结果同样证实了以上观点,家族具有丰富的多态性位点和遗传多样性,该家族在启动子区和CDS区存在多处点突变、插入和缺失现象,共鉴定出19种变异类型,其中有18种无毒功能丧失,另外1种是的突变类型,由于缺乏水稻抗性基因,因此还需要后续工作进一步验证此类型是否会引起无毒功能发生改变。转座子在启动子区或CDS区插入可导致新的毒力等位基因。Zhou等[43]在毒性野生型菌株B2中发现区域有转座子Pot3插入,导致菌株恢复毒性,但本研究所有供测菌株均未检测到转座子Pot3插入情况,与孟峰等[25]的研究结果一致。Dai等[44]报道了存在片段缺失和移码突变现象,共鉴定出27种变异类型。Chuma等[45]发现和在染色体上的位置及序列经常改变,但却能在7号染色体上以稳定形式存在,但本研究中,在多个位点发生单碱基替换,与前人研究不符,可能是由于稻瘟病菌与水稻协同进化过程中,水稻不断进化产生新的抗性基因,病原菌的无毒基因为了逃避抗性基因给予的选择压力,自身发生变异来克服小种抗性。
本研究还发现,不同地域来源的稻瘟病菌菌株群体遗传结构存在差异,与肖丹凤[46]对不同稻作区稻瘟病菌遗传分析的研究结果一致。Huang等[47]报道了中国不同群体的稻瘟病菌呈现出互相接近的遗传多样性,并且在地理位置上没有差异。但本研究中,黑龙江和海南稻作区内稻瘟病菌菌株群体之间的变异类型具有区域特征,即两稻区均含有其特异性等位基因型,与Zhang等[48]研究结果一致,中国南方和北方水稻产区支持两个截然不同的病原体群体,更加充分验证了不同稻区的稻瘟病菌群体间具有丰富的遗传多样性。这种特异性的存在,再一次证实了不同稻瘟病菌群体间的遗传特性与其所处的生态环境和耕作栽培等具有密切的相关性。
4 结论
家族在南北不同稻瘟病菌群体中均有分布且分布差异较大,并均含有其特异性变异类型。黑龙江菌株含有全部待测基因并均以高频率广泛存在;而海南菌株均以低频率集中并仅存在和,且黑龙江菌株较海南菌株无毒基因型组成种类丰富。同时家族在不同群体中均含有丰富的多态性和区域特征,两群体中家族均为突变后的等位基因型存在,共有19种变异类型,经致病性鉴定后,有18种失去无毒功能。检测出的所有突变类型均不能被相对应抗性基因和Pi-ta所识别,表现为感病。抗性基因和Pi-ta在黑龙江省和海南省对稻瘟病的抗病育种与利用过程中可聚合其他抗性基因应用来保证品种抗病性。因此,不同群体中稻瘟病菌复杂多样,应继续监测稻瘟病菌的变化情况,持续探究其在不同群体中的变异规律,及时掌握区域内田间稻瘟病菌动态,不仅对研究不同稻瘟病菌群体遗传结构的多样性具有重要意义,同时还可为区域内抗病种质的筛选与选育提供参考。
[1] HAYASHI K, YASUDA N, FUJITA Y, KOIZUMI S, YOSHIDA H. Identification of the blast resistance genein rice cultivars using functional markers. Theoretical and Applied Genetics, 2010, 121(7): 1357-1367.
[2] 张亚玲, 高清, 赵羽涵, 刘瑞, 付忠举, 李雪, 孙宇佳, 靳学慧. 黑龙江省水稻种质稻瘟病抗性评价及抗瘟基因结构分析. 中国农业科学, 2022, 55(4): 625-640.
ZHANG Y L, GAO Q, ZHAO Y H, LIU R, FU Z J, LI X, SUN Y J, JIN X H. Evaluation of rice blast resistance and genetic structure analysis of rice germplasm in Heilongjiang province. Scientia Agricultura Sinica, 2022, 55(4): 625-640. (in Chinese)
[3] FLOR H H. Current status of the gene-for-gene concept. Annual Review of Phytopathology, 1971, 9: 275-296.
[4] AZIZI P, RAFII M, ABDULLAH S, NEJAT N, MAZIAH M, HANAFI M, LATIF M, SAHEBI M. Toward understanding of rice innate immunity against. Critical Reviews in Biotechnology, 2016, 36(1): 165-174.
[5] 毛洧, 陈学伟, 王静. 水稻抗稻瘟病机制的研究进展. 中国科学: 生命科学, 2022, 52(10): 1495-1510.
MAO W, CHEN X W, WANG J. Recent progress on rice resistance to blast disease. Scientia Sinica Vitae, 2022, 52(10): 1495-1510. (in Chinese)
[6] 吴伟怀, 王玲, 程贯忠, 朱有勇, 潘庆华. 稻瘟病菌群体的分子遗传学研究——广东省与云南省稻瘟病菌群体遗传及致病型结构的比较分析. 中国农业科学, 2004, 37(5): 675-680.
WU W H, WANG L, CHENG G Z, ZHU Y Y, PAN Q H. Studies on molecular genetics of rice blast fungus population——comparison of genetic and pathotypic structures of two rice blast fungus populations derived from Guangdong and Yunnan provinces of China. Scientia Agricultura Sinica, 2004, 37(5): 675-680. (in Chinese)
[7] ORBACH M J, FARRALL L, SWEIGARD J A, CHUMLEY F G, VALENT B. A telomeric avirulence gene determines efficacy for the rice blast resistance gene. The Plant Cell, 2000, 12(11): 2019-2032.
[8] Kang S, Sweigard J A, Valent B. Thehost specificity gene family in the blast fungus. Molecular Plant-Microbe Interactions, 1995, 8(6): 939-948.
[9] SWEIGAED J A, CARROLL A M, KANG S, FARRALL L, CHUNLEY F G, VALENT B. Identification, cloning, and characterization of, a gene for host species specificity in the rice blast fungus. The Plant Cell, 1995, 7(8): 1221-1233.
[10] SCHNEIDER D R S, SARAIVA A M, AZZONI A R, MIRANDA H R, de Toledo M A S, PELLOSO A C, SOUZA A P. Overexpression and purification of, a mutant of the effector proteinfrom. Protein Expression and Purification, 2010, 74(1): 24-31.
[11] COLLEMARE J, PIANFETTI M, HOULLE A E, MORIN D, CAMBORDE L, GAGEY M J, BARBISAN C, FUDAL I, LEBRUN M H, BOHNERT H U.avirulence genebelongs to an infection-specific gene cluster involved in secondary metabolism. New Phytologist, 2008, 179(1): 196-208.
[12] FARMAN M L, LEONG S A. Chromosome walking to theavirulence gene of: discrepancy between the physical and genetic maps. Genetics, 1998, 150(3): 1049-1058.
[13] LI W, WANG B H, WU J, LU G D, HU Y J, ZHANG X, ZHANG Z G, ZHAO Q, FENG Q, ZHANG H Y, WANG Z Y, WANG G L, HAN B, WANG Z H, ZHOU B. Theavirulence geneencodes a predicted secreted protein that triggers the immunity in rice mediated by the blast resistance gene. Molecular Plant-Microbe Interactions, 2009, 22(4): 411-420.
[14] YOSHIDA K, SAITOH H, FUJISAWA S, KANZAKI H, MATSUMURA H, YOSHIDA K, TOSA Y, CHUMA I, TAKANO Y, WIN J, KAMOUN S, TERAUCHI R. Association genetics reveals three novel avirulence genes from the rice blast fungal pathogen. The Plant Cell, 2009, 21(5): 1573-1591.
[15] WU J, KOU Y J, BAO J D, LI Y, TANG M Z, ZHU X L, PONAYA A, XIAO G, LI J B, LI C Y,. Comparative genomics identifies theavirulence effectorthat triggers-mediated blast resistance in rice. New Phytologist, 2015, 206(4): 1463-1475.
[16] ZHANG S L, WANG L, WU W H, HE L Y, YANG X F, PAN Q H. Function and evolution ofavirulence generesponding to the rice blast resistance gene. Scientific reports, 2015, 5: 11642.
[17] JIA Y, MCADAMS S A, BRYAN G T, HERSHEY H P, VALENT B. Direct interaction of resistance gene and avirulence gene products confers rice blast resistance. The EMBO Journal, 2000, 19(15): 4004-4014.
[18] KHANG C H, PARK S Y, LEE Y H, VALENT B, KANG S. Genome organization and evolution of theavirulence gene family in thespecies complex. Molecular Plant-Microbe Interactions, 2008, 21(5): 658-670.
[19] 刘松青. 水稻稻瘟病抗性基因Pi-ta的候选基因筛选与初步分析[D]. 武汉: 中南民族大学, 2019.
Liu S Q. Screening and preliminary analysis of the candidate genes for rice blast resistance genePi-ta[D]. Wuhan: South-Central University for Nationalities, 2019. (in Chinese)
[20] Bryan G T, Wu K S, Farrall L, Jia Y, Hershey H P, McAdams S A, Faulk K N, Donaldson G K, Tarchini R, Valent B. A single amino acid difference distinguishes resistant and susceptible alleles of the rice blast resistance gene. The Plant Cell, 2000, 12(11): 2033-2045.
[21] 甘玉姿, 肖贵, 邓启云, 吴俊, 柏斌, 卢向阳, 周波. 菲律宾稻瘟病菌生理小种中及其同源基因的序列与功能分析. 中国生物防治学报, 2018, 34(3): 488-498.
GAN Y Z, XIAO G, DENG Q Y, WU J, BAI B, LU X Y, ZHOU B. The sequence and function analysis ofgene family of rice blast fungus,in Philippines. Chinese Journal of Biological Control, 2018, 34(3): 488-498. (in Chinese)
[22] 余欢, 姜华, 王艳丽, 孙国昌. 无毒基因在不同寄主梨孢菌中的变异研究. 浙江农业学报, 2015, 27(8): 1414-1421.
YU H, JIANG H, WANG Y L, SUN G C. Variability of avirulence genes inisolates from different hosts. Acta Agriculturae Zhejiangensis, 2015, 27(8): 1414-1421. (in Chinese)
[23] 刘瑞, 赵羽涵, 付忠举, 顾欣怡, 王艳霞, 靳学慧, 杨莹, 吴伟怀, 张亚玲. 黑龙江省和海南省基因家族在稻瘟病菌中的分布及变异. 中国农业科学, 2023, 56(2): 264-274.
LIU R, ZHAO Y H, FU Z J, GU X Y, WANG Y X, JIN X H, YANG Y, WU W H, ZHANG Y L. Distribution and variation ofgene family in ricefrom Heilongjiang province and Hainan province. Scientia Agricultura Sinica, 2023, 56(2): 264-274. (in Chinese)
[24] 李焕宇, 付婷婷, 张云, 吕天佑, 李远, 徐秉良. 5种方法提取真菌基因组DNA作为PCR模板效果的比较. 中国农学通报, 2017, 33(16): 28-35.
LI H Y, FU T T, ZHANG Y, Lü T Y, LI Y, XU B L. Effect comparison of five methods to extract fungal genomic DNA as PCR templates. Chinese Agricultural Science Bulletin, 2017, 33(16): 28-35. (in Chinese)
[25] 孟峰, 张亚玲, 靳学慧. 黑龙江省稻瘟病菌无毒基因及其同源基因的检测与分析. 中国水稻科学, 2020, 34(2): 143-149.
MENG F, ZHANG Y L, JIN X H. Detection and analysis ofavirulent geneand its homologous genes in Heilongjiang province. Chinese Journal of Rice Science, 2020, 34(2): 143-149. (in Chinese)
[26] SHI N N, RUAN H C, LIU X Z, YANG X J, DAI Y L, GAN L, CHEN F R, DU Y X. Virulence structure ofpopulations from Fujian province, China. Canadian Journal of Plant Pathology, 2018, 40(4): 542-550.
[27] 靳学慧. 农业植物病理学. 赤峰: 内蒙古科学技术出版社, 1999.
JIN X H. Agricultural Plant Pathology. Chifeng: Inner Mongolia Science and Technology Press, 1999. (in Chinese)
[28] 李思博, 魏松红, 王海宁, 罗文芳, 张优, 刘志恒. 2015-2016年辽宁省稻瘟病菌种群动态分析. 沈阳农业大学学报, 2017, 48(3): 284-289.
LI S B, WEI S H, WANG H N, LUO W F, ZHANG Y, LIU Z H. Dynamics of rice blast fungus population in Liaoning province in 2015-2016. Journal of Shenyang Agricultural University, 2017, 48(3): 284-289. (in Chinese)
[29] Valent B, Khang C H. Recent advances in rice blast effector research. Current Opinion in Plant Biology, 2010, 13(4): 434-441.
[30] 廖静静, 谢华, 王殿东, 何霞红. 云南省元阳县籼/粳型稻瘟病菌无毒基因、和多样性分析. 植物保护学报, 2019, 46(5): 1057-1064.
LIAO J J, XIE H, WANG D D, HE X H. Polymorphism analysis of,andin indica/japonica-borneisolates in Yuanyang county, Yunnan. Journal of Plant Protection, 2019, 46(5): 1057-1064. (in Chinese)
[31] 房文文. 稻瘟菌群体无毒基因型的时空动态研究[D]. 北京: 中国农业大学, 2018.
Fang W W. Temporal and spatial dynamics of avirulence genotypes in the populations of[D]. Beijing: China Agricultural University, 2018. (in Chinese)
[32] 王世维, 郑文静, 赵家铭, 魏松红, 王妍, 赵宝海, 刘志恒. 辽宁省稻瘟病菌无毒基因型鉴定及分析. 中国农业科学, 2014, 47(3): 462-472.
WANG S W, ZHENG W J, ZHAO J M, WEI S H, WANG Y, ZHAO B H, LIU Z H. Identification and analysis ofavirulence genes in Liaoning province. Scientia Agricultura Sinica, 2014, 47(3): 462-472. (in Chinese)
[33] 周江鸿, 王久林, 蒋琬如, 雷财林, 凌忠专. 我国稻瘟病菌毒力基因的组成及其地理分布. 作物学报, 2003, 29(5): 646-651.
ZHOU J H, WANG J L, JIANG W R, LEI C L, LING Z Z. Virulence genes diversity and geographic distribution ofin China. Acta Agronomica Sinica, 2003, 29(5): 646-651. (in Chinese)
[34] 高清, 张亚玲, 周弋力, 于连鹏, 聂强, 靳学慧. 黑龙江省粳稻品种稻瘟病主效抗性基因鉴定与抗性评价. 作物杂志, 2021(4): 59-66.
GAO Q, ZHANG Y L, ZHOU Y L, YU L P, NIE Q, JIN X H. Identification of major resistance genes and resistance evaluation to rice blast in japonica rice varieties in Heilongjiang province. Crops, 2021(4): 59-66. (in Chinese)
[35] 相亚超, 王丽丽, 徐凡, 马殿荣. 抗稻瘟病基因在黑龙江水稻资源中的分布. 分子植物育种, 2018, 16(23): 7705-7717.
XIANG Y C, WANG L L, XU F, MA D R. Study on the distribution of rice blast resistant genes in rice resources of Heilongjiang province. Molecular Plant Breeding, 2018, 16(23): 7705-7717. (in Chinese)
[36] 唐雪婷. 黑龙江省四个稻瘟病流行生态区稻瘟病菌无毒基因动态分析[D]. 大庆: 黑龙江八一农垦大学, 2021.
TANG X T. Dynamic analysis of avirulence genes ofin four epidemic ecological regions of Heilongjiang province[D]. Daqing: Heilongjang Bayi Agricultural University, 2021. (in Chinese)
[37] 董丽英, 王群, 刘树芳, 郑凤萍, 李迅东, 杨勤忠. 云南省稻瘟病菌群体对稻瘟病抗性单基因系的致病性分析. 西南农业学报, 2012, 25(2): 467-473.
DONG L Y, WANG Q, LIU S F, ZHENG F P, LI X D, YANG Q Z. Pathogenicity analysis ofpopulations of Yunnan on monogenic lines for resistance to rice blast. Southwest China Journal of Agricultural Sciences, 2012, 25(2): 467-473. (in Chinese)
[38] 曹雪琦. 福建省稻瘟病菌田间种群无毒基因的变异检测[D]. 福建: 福建农林大学, 2020.
CAO X Q. Detection of avirulence gene mutations in the field population of rice blast flungus in Fujian province[D]. Fujian: Fujian Agriculture and Forestry University, 2020. (in Chinese)
[39] 周瑚, 任佐华, 王恒沪, 张译允, 邹秋霞, 刘二明. 湖南桃江病圃稻瘟病菌的无毒基因及水稻抗瘟单基因联合抗性分析. 微生物学通报, 2017, 44(10): 2353-2360.
ZHOU H, REN Z H, WANG H H, ZHANG Y Y, ZOU Q X, LIU E M. Analysis of avirulence genes ofand resistance association of monogene against blast from rice blast nursery in Hunan Taojiang. Microbiology China, 2017, 44(10): 2353-2360. (in Chinese)
[40] 姜华, 余欢, 王艳丽, 孙国昌. 稻瘟病菌无毒基因序列变异研究进展. 浙江农业学报, 2015, 27(3): 512-520.
JIANG H, YU H, WANG Y L, SUN G C. Progress on sequence variation of avirulence genes in the rice blast fungus. Acta Agriculturae Zhejiangensis, 2015, 27(3): 512-520. (in Chinese)
[41] Sirisathaworn T, Srirat T, Longya A, Jantasuriyarat C. Evaluation of mating type distribution and genetic diversity of threeavirulence genes,,and, in Thailand rice blast isolates. Agriculture and Natural Resources, 2017, 51(1): 7-14.
[42] Kasetsomboon T, Kate-Ngam S, Sriwongchai T, Zhou B, Jantasuriyarat C. Sequence variation of avirulence gene. Mycological Progress, 2013, 12(4): 617-628.
[43] Zhou E, Jia Y, Singh P, Correll J C, Lee F N. Instability of theavirulence genealters virulence. Fungal Genetics and biology, 2007, 44(10): 1024-1034.
[44] Dai Y, Jia Y, Correll J, Wang X, Wang Y. Diversification and evolution of the avirulence gene. Fungal Genetics and Biology, 2010, 47(12): 973-980.
[45] Chuma I, Isobe C, Hotta Y, Ibaragi K, Futamata N, Kusaba M, Yoshida K, Terauchi R, Fujita Y, Nakayashiki H, VALENT B, TOSA Y. Multiple translocation of theeffector gene among chromosomes of the rice blast fungusand related species. PLoS Pathogens, 2011, 7(7): e1002147.
[46] 肖丹凤. 三个不同稻作区稻瘟病菌致病性与品种互作研究[D]. 北京: 中国农业科学院, 2013.
XIAO D F. Studies on the pathogenicity ofin three different rice growing regions and their interactions to rice varieties[D]. Beijing: Chinese Academy of Agricultural Sciences, 2013. (in Chinese)
[47] Huang J, Si W, Deng Q, Li P, Yang S. Rapid evolution of avirulence genes in rice blast fungus. BMC Genetics, 2014, 15: 45.
[48] ZHANG Y L, ZHU Q L, YAO Y X, ZHAO Z H, CORRELL J C, WANG L, PAN Q H. The race structure of the rice blast pathogen across southern and northeastern China. Rice, 2017, 10(1): 46.
Distribution and Variation Analysis offamily infrom Heilongjiang Province and Hainan Province

1College of Agronomy, Heilongjiang Bayi Agricultural University/Heilongjiang Plant Resistance Research Center, Daqing 163319, Heilongjiang;2Environment and Plant Protection Institute, Chinese Academy of Tropical Agricultural Sciences, Haikou 571101
【Objective】By detecting the distribution and variation characteristics of thefamily in differentstrains from Heilongjiang Province and Hainan Province, the pathogenic phenotypes of the variation types were understood, which provided a reference for the screening and breeding of disease-resistant germplasm in the region.【Method】Six pairs of specific primers were designed for the promoter region and CDS region by referring to thefamily sequence published in NCBI. DNA was extracted from 397 single spore strains ofcollected from different regions of Heilongjiang Province and Hainan Province in 2020, and PCR amplification was performed. By electrophoresis test results, representative strains from different regions were selected to sequence the amplified fragments. The sequencing results were compared with the corresponding base and amino acid sequences in NCBI, and the avirulence function of different variation types ofstrains was verified by using rice resistant single gene lines.【Result】In the PCR electrophoresis results, thestrains in Heilongjiang Province carried all the genes to be tested, which were widely distributed and had a high detection frequency. However, onlyandwere carried bystrains in Hainan Province, and concentrated in low frequency. The results of avirulence gene composition analysis showed that the strains ofin Heilongjiang were complex and diverse, and the genotypes were more abundant than those in Hainan. The sequencing results of PCR products showed that thefamily was divided into 19 types with point mutation, insertion and deletion as the main variation types, and the variation types of strains from differentpopulations were specific. Ten variation types were detected in, among which- (1-5) was a unique variation type ofstrains in Heilongjiang Province, and- (6-10) was a unique variation type ofstrains in Hainan Province. After functional verification of these ten variation types, it was found that the avirulence functions were lost. Eight variation types were detected in, among which- (1-4) was a unique variation type ofstrains in Heilongjiang Province, and- (5-8) was a unique variation type ofstrains in Hainan Province. After functional verification, it was found that the avirulence functions were lost. Only one variation type ()was detectedin【Conclusion】Thefamily in Heilongjiang and Hainan populations were mutated alleles. After pathogenicity identification, all mutation types could not be identified by the corresponding resistance genesandPi-ta. Therefore, the resistance genesandPi-tacan be used to polymerize other resistance genes to ensure the disease resistance of varieties in the process of disease resistance breeding and utilization of rice blast in Heilongjiang Province and Hainan Province. At the same time, the distribution and variation types offamily instrains from different geographical sources are specific.
;family; mutation; avirulence gene; Heilongjiang province; Hainan province

10.3864/j.issn.0578-1752.2023.03.006
2022-09-28;
2022-11-01
国家自然科学基金(U20A2025)、黑龙江省农垦总局科技攻关项目(HKKYZD190205)、黑龙江八一农垦大学科研启动项目(XDB201802,XDB201605)
刘瑞,E-mail:2271031165@qq.com。通信作者张亚玲,E-mail:byndzyl@163.com
(责任编辑 岳梅)
