Modulation of polymerization rate of N-carboxyanhydrides in a biphasic system
2023-02-18GuonnJiXuetoZhengXingieHouXioSunShijieWngXiohongLiJinjunChengbZiyunSong
Guonn Ji ,Xueto Zheng ,Xingie Hou ,Xio Sun ,Shijie Wng ,Xiohong Li ,Jinjun Chengb,,Ziyun Song,∗
a Institute of Functional Nano & Soft Materials (FUNSOM),Jiangsu Key Laboratory for Carbon-Based Functional Materials and Devices,Soochow University,Suzhou 215123,China
b Research Center for Industries of the Future,Westlake University,Hangzhou 310030,China
c School of Engineering,Westlake University,Hangzhou 310030,China
d The Key Lab of Health Chemistry and Molecular Diagnosis of Suzhou,College of Chemistry,Chemical Engineering and Materials Science,Soochow University,Suzhou 215123,China
Keywords:N-Carboxyanhydrides Polypeptides Biphasic system Cooperative covalent polymerization Polymerization kinetics
ABSTRACT The recent advances in accelerated polymerization of N-carboxyanhydrides (NCAs) offer an effective strategy to simplify the preparation of polypeptide materials.However,the fine-tuning of polymerization kinetics,which is critical to differentiate the main polymerization and the side reactions,remains largely unexplored.Herein we report the modulation of polymerization rate of NCA in a water/oil biphasic system.By altering the aqueous pH,the initial location of the initiators,and the pKa of initiating amines,we observed the change in polymerization time from several minutes to a few hours.Due to the high interfacial activity and low pKa value,controlled polymerization was observed from multi-amine initiators even if they were initially located in the aqueous phase.This work not only improves our understanding on the biphasic polymerization mechanism,but also facilitates preparation of versatile polypeptide materials.
Polypeptides,as the synthetic analogue of natural proteins,are one of the most important biomaterials because of their excellent biocompatibility,rich side-chain diversity,and the ability to adopt ordered secondary structures [1–7].Polypeptide materials have been widely studied in various biomedical fields such as drug delivery,gene delivery,tissue engineering,and antimicrobial applications [3,4,6,8–15].Despite the promising performance of polypeptide materials,the conventional preparation through the ring-opening polymerization (ROP) ofN-carboxyanhydride (NCA)monomers is limited by various side reactions,including the degradation of monomers,chain transfer,and chain termination,which significantly increase the cost and difficulty to obtain polypeptides in a controlled manner [16–19].
The recent advances in fast NCA polymerization offer an effective strategy to prepare versatile polypeptide materials [20–34],as accelerated polymerization outpaces various side reactions.We previously reported the development of an accelerated,cooperative covalent polymerization (CCP) of NCA in a water/oil biphasic system [26,35-37].The biphasic CCP allowed for the controlled synthesis of polypeptides in the presence of aqueous phase,which was otherwise impossible due to the hydrolysis of the monomers.The rate difference in desired polymerization and undesired waterinduced monomer degradation guaranteed that more than 99.9%monomer was consumed by the added initiators [26].Additionally,taking advantage of the fast polymerization kinetics,the biphasic CCP system was used to minimize the chain transfer and chain termination for the preparation of multiblock copolypeptides.The end-group fidelity of polypeptides remained>97% even after the synthesis of 20-block copolypeptides [35].Therefore,it is critical to manipulate the kinetics of biphasic CCP in order to differentiate it from various side reactions.Unfortunately,there are few reports to elucidate the impact of experimental conditions on the polymerization rate of biphasic CCP.
Here,we report the manipulation of the polymerization rate of biphasic CCP by tuning the aqueous pH,the initial location of the initiators,and the pKaof initiating amines (Fig.1).The decrease in aqueous pH or the partition of initiators into aqueous phase led to the protonation of propagating polypeptide chains,resulting in slower polymerization kinetics.Interestingly,the polymerization of NCA from multi-amine macroinitiators with interfacial activity and low pKavalue proceeded in an accelerated manner even if the macroinitiators were initially located in the aqueous phase.Therefore,polypeptides with branched architectures,including brush polymers entirely based on polypeptides,can be effi-ciently prepared by the biphasic CCP.We believe this work will help elucidate the mechanism of biphasic CCP,enabling the effi-cient preparation of various polypeptide materials by minimizing side reactions.
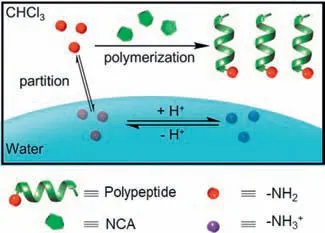
Fig.1. Scheme illustrating the modulation of biphasic CCP kinetics.
We have previously reported the development of an amphiphilic macroinitiator,methoxy poly(ethylene glycol)-blockpoly(γ-benzyl-L-glutamate) amine (PEG-PBLG),which enabled fast NCA polymerization in a water/dichloromethane (DCM) biphasic system [26].A polymerization at [M]0/[I]0=100 in the presence of an aqueous buffer (pH 7.0,1.0 wt%) finished within 20 min,outpacing water-induced degradation of NCA monomers.Since it is wellknown that the protonation of growing polypeptides slows down the polymerization [38,39],the biphasic CCP represents an interesting example that the propagating amino groups remain nonprotonated and nucleophilic even though the aqueous pH is lower than its pKa(∼8.0) [40].We reasoned that the interfacial anchoring of PEG-PBLG macroinitiators helped the orientation of PBLG block toward the DCM phase,which minimized the exposure of propagating amino groups to the aqueous phase and avoided the protonation.Such interfacial anchoring was validated by the molecular dynamic simulations in our previous studies [26].
To test the robustness of interfacially anchored PEG-PBLG macroinitiator against amine protonation,the polymerization kinetics ofγ-benzyl-L-glutamate NCA (BLG-NCA) from PEG-PBLG was monitored by1H NMR at even lower aqueous pH values of pH 3.0 and 5.0 in a water/DCM biphasic system ([M]0=0.1 mol/L,[M]0/[I]0=50,water:DCM=1:100 (w/w)) (Fig.2a).While the PEG block offered the interfacial anchoring of the macroinitiator,theαhelical PBLG segment guaranteed the one-stage kinetics by skipping the slower,random-coiled stage (Fig.S1 in Supporting information) [26].The polymerization kinetics became significantly slower as the aqueous pH decreased (Fig.2b and Fig.S2 in Supporting information),suggesting that the aqueous phase still participate in the polymerization process.Nevertheless,the NCA monomer was readily consumed even at pH 3.0,with>95% conversion in 130 min (Fig.2b),indicating that most of the propagating amines remained active and nucleophilic.While previous reports have shown that the protonated ammonium salt was able to polymerize NCA [39,41,42],the polymerization rate was slow that might not compete with the water-induced NCA degradation in a biphasic system.We therefore characterized the resulting polymersviagel permeation chromatography (GPC) to check the control over molecular weights (MWs).Interestingly,GPC results revealed monomodal peaks with low dispersity (Ð=Mw/Mn=1.05)for the polymers tested at all aqueous pH (Fig.2c and Table 1).The obtained MWs were similar among all tested conditions with aqueous pH 3.0,5.0,and 7.0 (Table 1),suggesting minimal water-induced degradation of monomers.The hydrolysis kinetics of NCA in a water/DCM emulsion further supported our judgement that CCP outpaced the side reactions (Fig.S3 in Supporting information).As the aqueous pH decreased,not only the biphasic CCP but also the NCA hydrolysis exhibited a slower rate,leading to controlled polypeptide synthesis from biphasic CCP at all aqueous pH.The results also validated our design to use biphasic CCP system to directly polymerize non-purified NCAs [36],as the segregation of impurities and the acidification of aqueous phase did not significantly alter the GPC results.
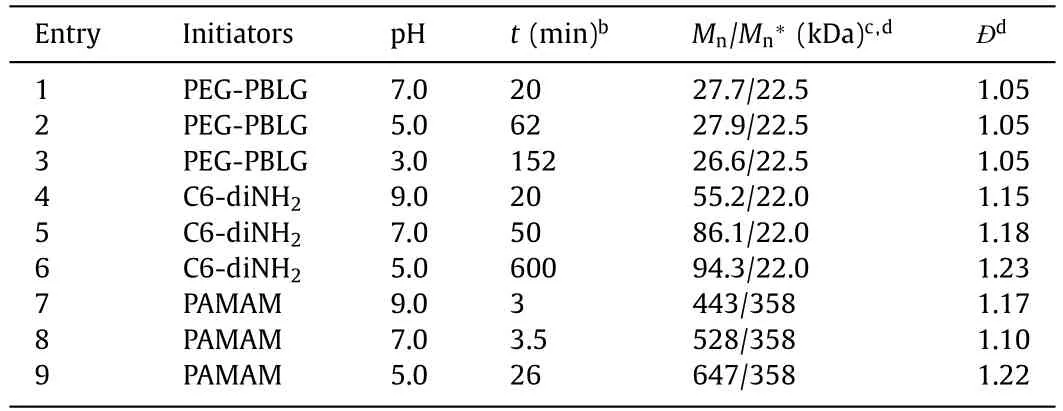
Table 1 Polymerization behavior in a biphasic system in the presence of different initiators at various aqueous pH.a

Fig.2. Biphasic CCP initiated from interfacially anchored PEG-PBLG.(a) Biphasic CCP of BLG-NCA initiated from PEG-PBLG.(b) Conversion of BLG-NCA in a water/CD2Cl2 biphasic system at various aqueous pH in the presence of PEG-PBLG.[M]0=0.1 mol/L,[M]0/[I]0=50,water:CD2Cl2=1:100 (w/w).(c) GPC traces of resulting polymers obtained from PEG-PBLG-initiated biphasic CCP at various aqueous pH.(d) Schematic illustration showing the reversible deactivation of PEG-PBLG propagating chains by the protonation of amino groups.
The pH-dependent kinetics likely originated from the non-zero solubility of water molecules in DCM [43].The diffusion of a few hydronium ions or other acidic species into the DCM phase resulted in partial,reversible protonation of the propagating amines(Fig.2d) [44].Therefore,the equilibrium between protonated and non-protonated propagating chain ends guaranteed simultaneous chain growth that led to well-defined polypeptides,albeit in an overall slower rate at acidic aqueous pH (i.e.,with more acidic species partitioned into the DCM phase).
The polymerization behavior of PEG-PBLG suggested that the location of initiators/propagating chains played an important role in the polymerization kinetics,as the partition of propagating amines into the aqueous phase may lead to their protonation and deactivation.We therefore continued to check the CCP from 1,6-hexanediamine (C6-diNH2) in the water/chloroform biphasic system (Fig.3a).C6-diNH2exhibited rapid polymerization kinetics compared with its mono-amine (i.e.,n-hexylamine) analogue [25].It was selected due to the good solubility in both aqueous and organic phase,which allowed us to investigate the impact of initial location of initiators on the polymerization profile (Fig.3b).We first dissolved C6-diNH2in aqueous buffer at various pH,and then added the aqueous solution of C6-diNH2into a chloroform solution of BLG-NCA to start the polymerization ([M]0=0.1 mol/L,[M]0/[I]0=50,water:chloroform=1:100 (w/w)) (Fig.3c).As shown in Fig.3c,the polymerization exhibited an even stronger dependence on the aqueous pH.Fast polymerization of NCA was observed at pH 9.0,reaching>92% conversion within 22 min.The decrease in aqueous pH significantly slowed down the polymerization rate.While the polymerization at an aqueous pH 7.0 still proceeded in a reasonably fast manner (∼84% conversion after 44 min),the consumption of NCA monomer was only 25% after 75 min at an aqueous pH of 5.0.
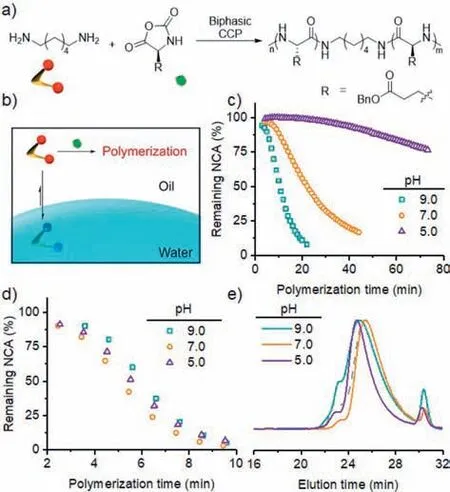
Fig.3. Biphasic CCP in the presence of C6-diNH2.(a) Biphasic CCP of BLG-NCA initiated from C6-diNH2.(b) Schematic illustration showing the partition and protonation of C6-diNH2.Conversion of BLG-NCA in a water/CDCl3 biphasic system at various aqueous pH in the presence of C6-diNH2,which was initially dissolved in the aqueous phase (c) or CDCl3 phase (d).[M]0=0.1 mol/L,[M]0/[I]0=50,water:CDCl3=1:100 (w/w).(e) GPC traces of resulting polypeptides obtained from C6-diNH2-initiated biphasic CCP at various aqueous pH,with C6-diNH2 initially dissolved in the organic phase.Polypeptide initiated by C6-diNH2 in a solution polymerization in chloroform (gray dash line) was used as a reference.
With the slower polymerization rate when C6-diNH2was initially dissolved in the aqueous phase,it was possible that the water-induced side reactions could not be completely inhibited,leading to competing initiation of NCA monomers.Indeed,the consumption of monomers in the absence of C6-diNH2was comparable to that in the presence of C6-diNH2at all three aqueous pH values in the water/chloroform biphasic system (Fig.S4 in Supporting information),suggesting that the obtained polypeptides were at least partially initiated by water molecules at the interface.While the NCA consumption was still faster with the addition of C6-diNH2at pH 7.0 and 9.0,the NCA conversion was almost identical at pH 5.0 with and without C6-diNH2.In order to check the competition of initiation between C6-diNH2and water,the obtained polypeptides in the presence and absence of C6-diNH2were characterized by GPC.The MWs were significantly smaller in the presence of C6-diNH2compared to that in the absence of C6-diNH2at pH 9.0 (Fig.S5 in Supporting information),suggesting the participation of C6-diNH2in the initiation process.In contrast,there were negligible differences between the MWs in the presence and absence of C6-diNH2at pH 7.0 (Fig.S5),indicating insignificant initiation from C6-diNH2at lower pH.While C6-diNH2contributed to the initiation at pH 9.0,we could not tune the MWs by varying the amount of C6-diNH2.GPC analysis revealed that the MWs of resulting PBLG was independent of the amount of C6-diNH2that was initially added into the aqueous phase at all tested conditions(Table 1,Table S1 and Fig.S6 in Supporting information).Compared to the PEG-PBLG macroinitiator,C6-diNH2lacked sufficient interfacial activity to stay at the water/oil interface.As a result,it was likely that the initiating amines became protonated in the aqueous phase and failed to rapidly polymerize NCA monomers in the chloroform phase (Fig.3b).
In sharp contrast to the biphasic CCP with water-dissolved C6-diNH2,the polymerization kinetics was independent of the aqueous pH when C6-diNH2was initially located in chloroform (Fig.3d).The polymerization rates under all three tested conditions (i.e.,aqueous pH 5.0,7.0,and 9.0) were rapid,with>99% monomers consumed within 15 min.Because of the fast polymerization even at the initial stage (monomer conversion ∼10% in the first 2.5 min),the C6-diNH2initiator quickly grew into oligomeric PBLG chains,which exhibited poor solubility and tend to stay in the chloroform phase (Fig.S7 in Supporting information).The fast polymerization kinetics ensured that the obtained polypeptides were initiated by C6-diNH2initiators.The GPC traces of resulting polypeptides in all three conditions showed great resemblance with each other as well as that obtained from the solution polymerization in chloroform (Fig.3e and Table S2 in Supporting information),suggesting that water-induced monomer degradation was minimal.Matrix-assisted laser desorption ionization-time of flight (MALDITOF) mass spectrometry confirmed that only C6-diNH2-initiated polypeptides were observed (Fig.S8 in Supporting information).The larger MWs than expected values and the broad molecular weight distribution (MWD) resulted from the two-stage kinetics,where the folding of polypeptides intoα-helices accelerated the kinetics [21,27].Propagating chains entering the second stage thus outgrew those staying in the first stage,resulting in a mixture of longer polypeptides and shorter oligomers with DP<10.Indeed,a shoulder peak at the low MW region was observed on the GPC traces of all C6-diNH2-initiated polymerization (Fig.3e).Therefore,by changing the initial location of C6-diNH2,we were able to modulate the kinetics of biphasic CCP,hence controlling the competition between desired polymerization and undesired side reactions.
Besides the interfacial activity of the initiator and the aqueous pH,we hypothesized that the pKaof the initiator might be another critical structural factor to influence the biphasic CCP kinetics.The protonation of amino initiating sites in aqueous phase not only deactivated the propagating chains,but also slowed down the partitioning process into the organic phase.As a result,an amino initiator with a lower pKawould stay as the non-protonated form with a broader range of aqueous pH,facilitating the transfer into the oil phase and the subsequent polymerization.To validate our hypothesis,the pKaof C6-diNH2was determined by potentiometric method with a pH meter.The titration curve of C6-diNH2revealed a buffering region between pH 9.0–11.3,with the pKadetermined as 10.1 that was consistent with the literature value (∼11.0) (Fig.4) [45].Almost all amino groups became protonated when C6-diNH2was dissolved in the aqueous solution at pH 5.0,7.0,and 9.0.The charged,protonated form of C6-diNH2preferred to stay in the aqueous phase,exhibiting a much slower initiation that could not fully inhibit water-induced side reactions.
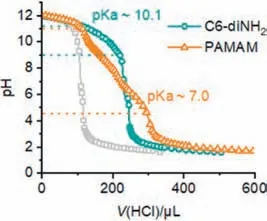
Fig.4. Titration curve of C6-diNH2 and PAMAM.The buffering region was indicated with dashed lines.The titration curve of water (gray lines) was used as a reference.
Therefore,an ideal initiator for biphasic CCP would not only show a high tendency to partition into the organic phase for polymerization,but also have a relatively low pKavalue to remain active even at neutral pH.With these requirements,multi-amine such as poly(amidoamine) (PAMAM) dendrimer serves as a promising candidate.Previous work reported the high interfacial activity of polyelectrolytes,which benefited the interfacial anchoring in biphasic CCP [46].Additionally,PAMAM (generation 3.0 with 32 amino groups) exhibited a broader range of buffering effect from pH 4.5 to 10.9,as evidenced by the titration curve (Fig.4).The pKaof PAMAM,which indicated the aqueous pH with 50% of the amino groups protonated,was determined to be 7.0 that agreed well with previous literature value (∼6.85) [47].Due to the formation of intramolecular hydrogen bonds after the ionization of several amino groups,it became more difficult to protonate the rest of amino groups [48].As a result,PAMAM exhibited a significantly lower pKacompared to a conventional primary amino group,with half of the amino groups remained non-protonated at pH 7.0.Besides the high interfacial activity and low pKavalue,the selection of PAMAM as biphasic initiator also helped minimize the side reactions due to the ultrafast kinetics [29].
The biphasic CCP kinetics was monitored at various aqueous pH when PAMAM was initially dissolved in either aqueous or chloroform phase (Fig.5a).When PAMAM macroinitiator was dissolved in chloroform and mixed with the biphasic mixture containing NCA,pH-independent,ultrafast polymerizations were observed with complete monomer conversion within 4.5 min,as monitored by FTIR (Fig.5b).The ultrafast polymerization rate guaranteed rapid growth of polypeptides that facilitated the partition of PAMAM in oil phase,outpacing water-induced degradation of NCA monomers.The resulting polypeptides from PAMAM exhibited similar MWs and MWDs among all three conditions with aqueous pH of 5.0,7.0,and 9.0 (Fig.S9 in Supporting information).

Fig.5. Biphasic CCP in the presence of PAMAM.(a) Biphasic CCP of BLG-NCA initiated from PAMAM.(b) FTIR spectra of polymerization mixture after 4.5 min incubation in a water/chloroform biphasic system at various aqueous pH in the presence of PAMAM,which was initially dissolved in the chloroform phase.[M]0=0.1 mol/L,[M]0/[I]0=50,water:chloroform=1:100 (w/w).(c) Conversion of BLG-NCA in a water/CDCl3 biphasic system at various aqueous pH in the presence of PAMAM,which was initially dissolved in the aqueous phase.(d) GPC traces of resulting polypeptides obtained from PAMAM-initiated biphasic CCP at various aqueous pH,with PAMAM initially dissolved in the aqueous phase.
In contrast to the biphasic CCP with chloroform-dissolved PAMAM,the polymerization initiated from water-dissolved PAMAM exhibited pH-dependence.While the CCP was still fast with an aqueous pH of 7.0 or 9.0,reaching>99% conversion within 10 min,the CCP at an aqueous pH of 5.0 was much slower that took∼30 min to finish (Fig.5c).FTIR characterization under continuous stirring conditions was consistent with NMR kinetics,where the biphasic CCP at pH 5.0 was significantly slower when PAMAM was initially dissolved in the aqueous phase (Fig.S10 in Supporting information).Therefore,when the aqueous pH was lower than the pKaof the amino initiators,the inhibitory effect of amine protonation was profound even though the initiators exhibited strong interfacial activity.While the GPC characterization revealed controlled polymerization at aqueous pH of 9.0 and 7.0,the analysis of resulting polypeptides at pH 5.0 showed a larger MW than expected value,presumably due to the loss of partial PAMAM macroinitiators in the aqueous phase (Fig.5d and Table 1).
It has to be noted that the dissolution of PAMAM in chloroform relied on the use of methanol,mainly because of the poor solubility of polar multi-amine in chloroform.As a result,conventional preparation of star-or brush-polypeptides was limited due to the incompatible solubility of multi-amine macroinitiator (i.e.,favors water as solvent) and NCA monomer (i.e.,disfavors water due to degradation).For instance,poly(L-lysine) (PLL) exhibited poor solubility in almost all organic solvents,which was regarded as an undesired macroinitiator for polypeptide synthesis that usually generated polypeptides with low degree of polymerization (DP) or broad MWD [49,50].Recent efforts have been made to derivatize the multi-amine initiators for better solubility and similar reactivity (e.g.using trimethylsilyl group) that complicated the synthetic process [21,51].Considering the high interfacial activity and fast NCA polymerization kinetics of multi-amines,biphasic CCP offered an effective strategy for the facile preparation of star-or brushpolypeptides by dissolving the macroinitiator in the aqueous phase with a proper pH (i.e.,larger than the pKa) [52].
To check the MW control of biphasic CCP from water-dissolved multi-amine initiator,PAMAM was dissolved in aqueous buffer at pH 7.0,which was mixed with the chloroform solution of BLGNCA at different [M]0/[I]0to initiate the polymerization.All polymerizations were completed (>99% conversion) within 6 min,outpacing NCA degradation in a water/chloroform mixture at pH 7.0(Fig.S4).GPC characterization suggested well-controlled polymerization with predictable MWs and low dispersity (Fig.6a and Table S3 in Supporting information).The slightly larger MW likely resulted from the loss of a small fraction of PAMAM initiators in the aqueous phase.Due to the rigid-rod conformation ofα-helices,the size of resulting PAMAM-PBLG unimolecular micelles should be proportional to the DP of PBLG [29].As expected,the dynamic light scattering (DLS) characterization of PAMAM-PBLG in DMF exhibited well-defined size distribution with low dispersity (Fig.6b and Table 2).The size of PAMAM-PBLG ranged from 28 nm to 64 nm,agreeing well with the expected value calculated from the DP (see Supporting information for details).In a similar strategy,the aqueous solution of PLL was used to prepare brush polymers entirely based on polypeptides (Scheme S1 in Supporting information).GPC characterization revealed a monomodal peak of the resulting PLLPBLG brush polypeptides with a narrow dispersity (Ð=1.15) (Fig.S11 in Supporting information),substantiating the use of biphasic CCP to prepare star-or brush-polypeptide materials with an aqueous solution of multi-amine initiators.

Table 2 Preparation of PAMAM-PBLG star polypeptides with biphasic CCP.a
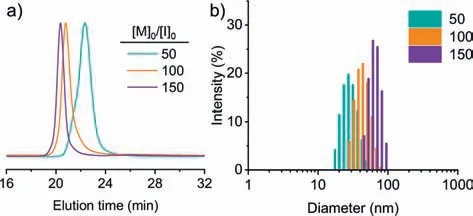
Fig.6. Preparation of star polypeptides by biphasic CCP.(a) GPC traces of resulting PAMAM-PBLG star polypeptides obtained from biphasic CCP at various [M]0/[I]0,with PAMAM macroinitiators initially dissolved in the aqueous phase.(b) DLS characterization of PAMAM-PBLG with different DP of polypeptides.
In summary,we reported the control over polymerization kinetics of biphasic CCP by altering the aqueous pH,the initial location of initiators,as well as the pKaof the initiating groups.With sufficient rate acceleration,water-induced degradation of NCA monomers was minimized even at basic pH,substantiating the robustness of biphasic CCP to prepare polypeptide materials.With the lower pKaand the interfacial activity of multi-amine initiators,star-and brush-polypeptides were efficiently synthesized even though the initiators were initially dissolved in the aqueous phase at neutral pH.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work is supported by the National Natural Science Foundation of China (No.22101194 for Z.Song and No.52233015 for J.Cheng),Natural Science Foundation of Jiangsu Province (No.BK20210733 for Z.Song),Suzhou Municipal Science and Technology Bureau (No.ZXL2021447 for Z.Song),Collaborative Innovation Center of Suzhou Nano Science &Technology,the 111 Project.,Joint International Research Laboratory of Carbon-Based Functional Materials and Devices,and Suzhou Key Laboratory of Nanotechnology and Biomedicine.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2023.108872.
杂志排行

Chinese Chemical Letters的其它文章
- Spin switching in corrole radical complex
- Benzothiadiazole-based materials for organic solar cells
- Mono-functionalized pillar[n]arenes: Syntheses,host–guest properties and applications✰
- Recent advances in two-step energy transfer light-harvesting systems driven by non-covalent self-assembly✩
- From oxygenated monomers to well-defined low-carbon polymers
- Doping-induced charge transfer in conductive polymers