基质相互作用分子1/钙通道蛋白1在哮喘气道高反应性中的上皮表达和作用
2023-02-12曾宪聪
桂 萍, 曾宪聪
(湖北省十堰市人民医院 呼吸内科, 湖北 十堰, 442000)
哮喘是全球最常见的慢性病之一[1]。气道高反应性(AHR)是过敏性哮喘的主要特征[2]。AHR与气道平滑肌(ASM)收缩有关,并受胞质钙离子(Ca2+)水平调节,突出了Ca2+稳态在哮喘中的重要性[2]。全基因组分析确定了12个与哮喘相关的表观遗传相互作用基因,包括基质相互作用分子1(STIM1)[3]。STIM1是一种钙调节激素,广泛分布于肾脏、子宫内膜和肺等组织中,可以通过激活钙通道蛋白1(Oria1)介导钙库操纵的钙内流(SOCE)[4]。因此,本研究假设STIM1可能是与哮喘AHR相关的重要上皮衍生因子,其通过介导Ca2+流入来调节ASM收缩。验证这一假设,本研究拟通过建立哮喘小鼠模型,探讨STIM1/Orail信号通路介导的SOCE在AHR中的作用。
1 材料与方法
1.1 实验动物
36只雌性Balb/c小鼠[8~10 周龄, (20.0±1.1) g]购自北京维通利华实验动物技术有限公司。小鼠在标准条件[温度为(22±1) ℃; 湿度为55%~60%; 12 h明暗循环)]下饲养,可自由获取水和食物。适应1周后,将小鼠随机分为3组(每组12只),即对照组(CON)、鸡卵清蛋白(OVA)组和OVA+sh-STIM1组(OVA来源于鸡蛋,是一种常用的过敏原,可在实验室啮齿动物中诱发过敏性肺部炎症。)。参照文献[5]中方法建立OVA诱导的小鼠急性哮喘模型。除对照组外,其他组分别在第0、7、14天通过腹腔注射50 μg OVA(纯度≥98%, 美国Sigma-Aldrich公司)致敏。小鼠用异氟醚麻醉,连续3 d(第21、22、23天)通过吸入溶解在100 μL生理盐水中的150 μg OVA进行致敏。对照组采用等量生理盐水处理。OVA+sh-STIM1组小鼠在第21、22、23天用OVA致敏前8 h, 通过鼻内给予100 μL慢病毒介导的STIM1小干扰RNA[sh-STIM1, 汉恒生物科技(上海)有限公司]。
1.2 慢病毒介导的短发夹载体的构建与感染
对于短发夹RNA (shRNA) 介导的STIM1敲低,用从上海GeneChem Corporation公司购买的STIM1-pGCSIL-GFP质粒产生的慢病毒颗粒转染细胞。shRNA的靶向序列为[6]: 5′-GGAGGATAA TGGCTCTATT-3′。阴性对照是与任何已知人类基因没有序列同源性的双链shRNA。对于基因沉默,将纯化的慢病毒(sh-STIM1-1)以20的感染复数添加到细胞中,持续8 h, 并用培养基洗涤2次。通过GFP表达监测发现感染复数为20时, 72 h后癌细胞感染率超过90%。因此,本研究在所有实验中对慢病毒使用了20的感染复数,因为其在所需的时间内产生了最佳的基因敲低。对照细胞用阴性对照shRNA感染。
1.3 气道高反应性的测量
在实验第24天,通过全身气压体积描记在8~10周龄的小鼠(每组6只小鼠)中评估对乙酰甲胆碱(Mch, 美国Sigma-Aldrich公司)的气道反应性。小鼠用戊巴比妥(100 mg/kg)腹腔注射麻醉,并暴露于含Mch(0、3.125、6.250、12.500、25.000和50.000 mg/mL)的磷酸盐缓冲液(PBS)雾化中,利用全身瀑体积测量法(型号 PLY 3211, 美国Buxco Electronic公司)记录增强的停顿(Penh)。
1.4 苏木精和曙红染色及炎症评分
在实验第24天,处死小鼠,取右上叶石蜡包埋,矢状切片(5 μm厚),苏木精-伊红(HE)染色。将炎症浸润程度定为: 0分,无炎症细胞; 1分,炎症细胞少; 2分,炎症细胞呈圆形,厚度为1个细胞; 3分,炎症细胞呈圆形,厚度为2~4个细胞; 4分,炎症细胞形成圆形,厚度>4个细胞。由对样本不知情的研究人员在每张载玻片上随机选择的6个区域中对炎症进行评分。总炎症评分计算为所有个体炎症评分的平均值。
1.5 免疫荧光染色
肺组织切片(5 μm) 进行抗原修复,并在室温下浸入山羊血清15 min, 以避免非特异性结合。然后将切片与1∶50稀释的抗STIM1 (美国Santa cruz公司) 一抗在4 ℃下孵育过夜。PBS洗涤3次后,将切片与1∶200稀释的Cy3标记的山羊抗兔IgG抗体(上海Beyotime公司)在室温下一起孵育90 min。最后,切片用二脒基苯基吲哚(DAPI)染色并用PBS洗涤3次。在放大倍数为400倍的荧光显微镜下观察切片。
1.6 免疫组织化学
通过免疫组织化学染色检测α-平滑肌肌动蛋白(α-SMA)在肺组织中的表达。用于免疫染色的主要抗体如下: 兔抗α-SMA多克隆抗体(1∶100, 英国Abcam公司)。二级抗体为HRP标记的抗兔抗体(美国Jackson ImmunoResearch公司)。
1.7 酶联免疫吸附试验(ELISA)
所有组均进行气管分离,行气管插管,用0.5 mL PBS灌洗小鼠支气管肺泡,收集支气管肺泡灌洗液(BALF), 共3次,每次回收率达82%。根据ELISA试剂盒说明(美国R&D公司),检测小鼠BALF中白细胞介素(IL)-4、IL-5、单核细胞趋化蛋白(MCP-1)和IL-13水平。
1.8 细胞培养
人气道平滑肌细胞(HASMC,美国Sciencell公司)接种在SMCM培养基(Sciencell)中,并在37 ℃、5%CO2的湿润环境中培养。HASMC连续传代并取第4代用于实验。将HASMC以1×104个细胞/cm2的密度接种在细胞培养板中。细胞实验分为4组: 对照组(CON)、ACh组、ACh+sh-STIM1组和sh-STIM1组。ACh+sh-STIM1组和sh-STIM1组细胞在乙酰胆碱(ACh)干预前用sh-STIM1-1感染细胞8 h,以敲低细胞中STIM1表达。
1.9 细胞收缩试验
将HASMC重新悬浮在使用冷胶原蛋白Ⅰ型(纯度=90%, 溶于0.02 mol/L乙酸的液体, 1.5 mg/mL, 美国BD Biosciences公司)制备的胶原蛋白溶液中,该胶原蛋白溶液每孔含有1.5×105个细胞。将凝胶加入24孔板 (600 μL/孔) 并在 37 ℃下聚合90 min。将ACh(美国Sigma-Aldrich公司)添加到适当的孔中至终浓度为100 μmol/L, 并在4 h时拍摄照片。处理后立即使用灭菌探针从孔边缘提起凝胶。使用NIH Image J 在每个时间点测量每个凝胶的表面积。
1.10 钙成像
通过Fura-2AM(钙离子荧光探针,上海Beyotime公司)测量HASMC中的钙成像。将24孔板(2.5×103个细胞/孔)中的HASMC与250 μL含有2.5 μL/mL Fura-2AM的标准林格氏溶液(pH值为7.4)孵育30 min。然后用HBSS洗涤细胞3次。将HBSS中的ACh(100 μmol/L)或thapsigargin抑制剂(TG, 2 μmol/L)添加到相关孔中,并在37 ℃下孵育2 h。去除上清液以拍摄图像。使用珀耳帖冷却电荷耦合器件(CCD)相机(美国Roper Scientific公司)在荧光显微镜(10倍物镜)下检测细胞中的Fura-2AM荧光。使用荧光滤光片组在495 nm和518 nm处交替获得Fura-2AM荧光。信号转换为相对变化(F/F0), 其中F0是时间0的平均荧光强度(495/518)的比率。每孔记录15个细胞。
1.11 蛋白质印迹
使用SDS裂解缓冲液收获细胞。不同处理组中等量的蛋白质进行10%十二烷基硫酸钠聚丙烯酰胺凝胶电泳(SDS-PAGE)并转移到硝酸纤维素膜(美国Millipore公司)。膜在3%牛血清白蛋白(BSA)中封闭,随后与兔多克隆Oria1抗体(1∶1 000, 美国Cell Signaling Technology公司)、兔单克隆抗体GAPDH(1∶1 000, 美国Cell Signaling Technology公司)在4 ℃下孵育过夜。HRP偶联的单克隆山羊抗兔IgG(1∶5 000, 上海Bioworld公司)抗体用作二抗。使用ECL试剂(美国Millipore公司)显示条带。使用NIH Image Pro对光密度扫描进行量化,并将值表示为与GAPDH相比的相对强度。
1.12 实时定量聚合酶链反应(PCR)
使用Trizol试剂(美国Invitrogen公司)分离总RNA。然后使用PrimeScriptTM逆转录酶(日本Takara公司)将1 μg总RNA逆转录cDNA, 并使用TaqMan Gene Expression Assay试剂盒在ABI Prism 7000(美国Applied Biosystems公司)上进行定量实时PCR分析。PCR中使用的引物如下,STIM1: 5′-AGTCACAGTGAGAAGGCGAC-3′(正向), 5′-CAATTCGGCAAAACTCTGCTG-3′(反向);Orai1: 5′-GACTGGATCGGCCAGAGTTAC-3′(正向), 5′-GTCCGGCTGGAGGCTTTAAG-3′(反向);GAPDH: 5′-GGAGCGAGATCCCTCCAAAAT-3′(正向), 5′-GGCTGTTGTCATACTTCTCATGG-3′(反向)。循环条件是在95 ℃下初始变性30 s, 然后在95 ℃ 5 s、60 ℃ 30 s和72 ℃ 10 min进行40个循环。使用2-△△CT方法计算数据并归一化为GAPDH。
1.13 统计学分析
2 结 果
2.1 STIM1在鼠支气管上皮细胞中高表达
OVA组小鼠表现出气道阻力增加(图1A)以及组织(图1B)和BALF(图1C、D)炎症。免疫荧光染色分析表明,与对照组相比,在OVA处理动物的支气管上皮细胞中STIM1表达增加(图1E、F), 差异有统计学意义(P<0.05)。这些结果表明STIM1在哮喘过敏模型中表达增加。

A: 气道阻力的评估,各组通过flexiVent测量对乙酰甲胆碱(0~50 mg/mL)剂量增加的气道阻力,以记录增强暂停(n=6); B: 气道炎症的评估,苏木精-伊红染色观察支气管周围和肺泡区域的炎性细胞浸润; C: 各组之间的BALF中总白细胞计数; D: OVA组和OVA+sh-STIM1组BALF中白细胞的数量(NEU: 中性粒细胞; LYM: 淋巴细胞; MON: 单核细胞; EOS: 嗜酸性粒细胞); E、F: 各组支气管上皮细胞中STIM1表达的免疫荧光染色分析。与对照组相比, *P<0.05, **P<0.01, ***P<0.001; 与OVA组相比, #P<0.05, ###P<0.001。图1 STIM1在鼠支气管上皮细胞中高表达
2.2 STIM1敲低消除过敏性哮喘小鼠OVA诱导的AHR和ASM收缩
为了确定STIM1水平的降低是否与过敏性哮喘的病理学相关,本研究探讨了STIM1敲低对AHR的影响(图1E、F)。与对照组相比, OVA组Mch激发后气道阻力增加,这被鼻内sh-STIM1逆转(图1A、B)。免疫组织化学分析表明, OVA增强α-SMA表达(ASM激活的标志物)被鼻内sh-STIM1显著抑制(图2)。这些结果表明STIM1在哮喘AHR中具有作用。
2.3 STIM1敲低抑制炎症细胞浸润及相关因子释放
OVA显著增强肺泡和支气管周围的炎性细胞浸润,鼻内sh-STIM1治疗显著减弱(图1B)。BALF细胞计数表明, sh-STIM1给药显著抑制BALF中的细胞总数以及嗜酸性粒细胞、中性粒细胞和巨噬细胞的数量(图1C、D)。OVA增强BALF表达Th2细胞因子释放,并且sh-STIM1治疗减弱OVA诱导的IL-13、IL-5和MCP-1释放,但对OVA诱导的IL-4释放无影响。见表1。
2.4 STIM1敲低对ASM细胞的收缩力和Ca2+流入影响
为了进一步探讨STIM1对ASM的影响,本研究在体外使用了HASMC。ACh(100 μmol/L)诱导胶原基质中的HASMC在4 h后收缩约80%, 而对照组收缩约40%。sh-STIM1对HASMC的收缩没有影响[(41.31±3.27)%与(44.49±0.36)%]。然而,与sh-STIM1预处理显著抑制ACh诱导的收缩,表明ACh通过降低ASM收缩性来调节AHR(图3A)。ASM收缩性由Oria1调节, ACh增加Oria1表达,通过sh-STIM1预处理完全减弱(图3B)。ACh以浓度依赖性的方式增加了HASMC的SOCE, 这被sh-STIM1预处理完全消除(图3C)。用非竞争性Ca2+ATP酶TG抑制剂(2.0 μmol/L)和1.2 mmol/L Ca2+处理HASMC可激活SOCE, 并进一步升高细胞质Ca2+水平,而sh-STIM1可降低这种效应(图3D)。PCR结果显示, TG增强了STIM1mRNA和Orai1mRNA在HASMC中的表达,而sh-STIM1抑制了STIM1和Orai1表达(图3E)。

与对照组相比, **P<0.01; 与OVA组相比, #P<0.05。图2 通过免疫组织化学定量支气管周围α-SMA蛋白表达和定量分析(n=6)

表1 STIM1敲低对BALF中炎症因子释放影响 pg/mL
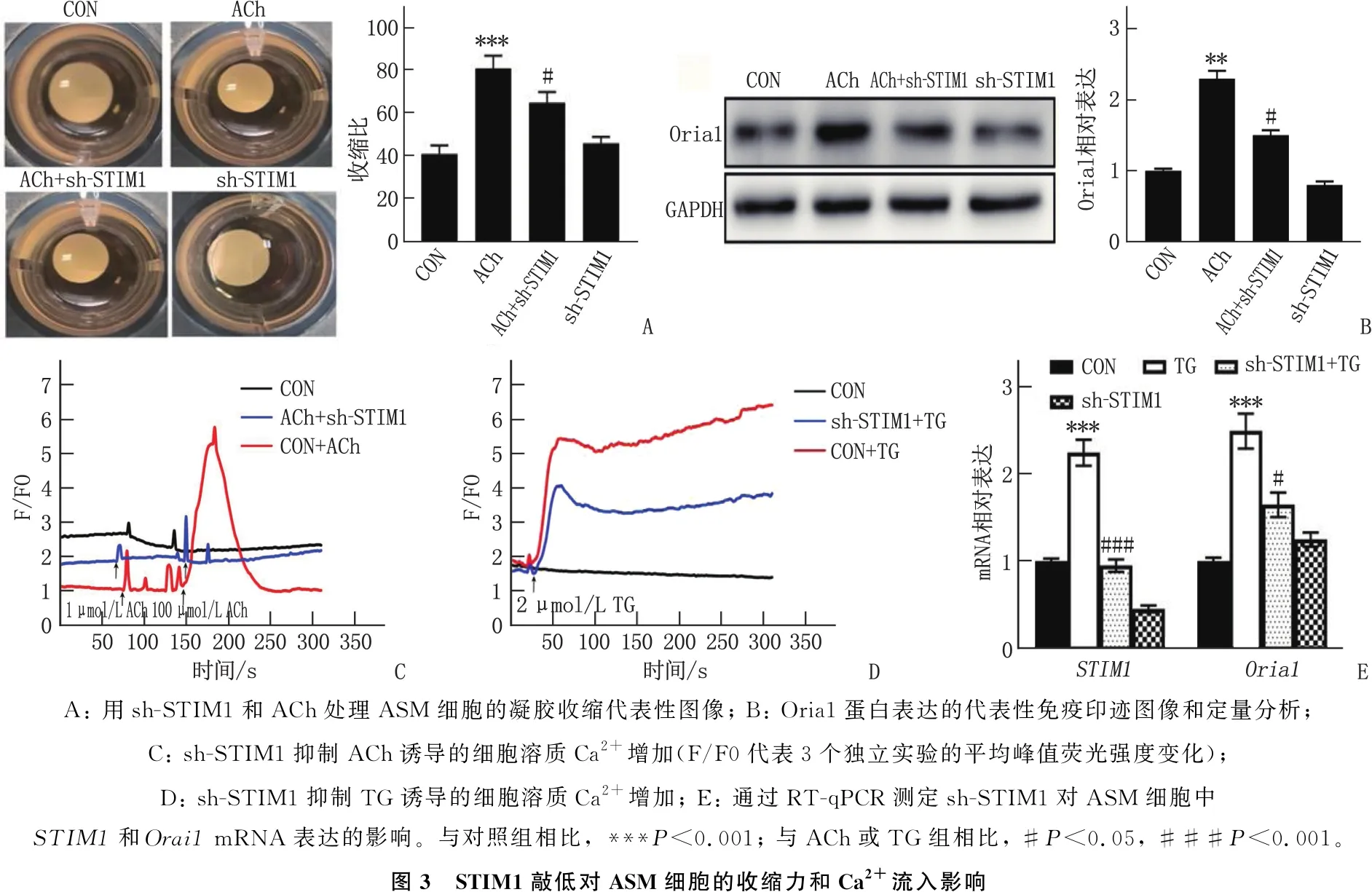
A: 用sh-STIM1和ACh处理ASM细胞的凝胶收缩代表性图像; B: Oria1蛋白表达的代表性免疫印迹图像和定量分析; C: sh-STIM1抑制ACh诱导的细胞溶质Ca2+增加(F/F0代表3个独立实验的平均峰值荧光强度变化); D: sh-STIM1抑制TG诱导的细胞溶质Ca2+增加; E: 通过RT-qPCR测定sh-STIM1对ASM细胞中STIM1和Orai1 mRNA表达的影响。与对照组相比, ***P<0.001; 与ACh或TG组相比, #P<0.05, ###P<0.001。图3 STIM1敲低对ASM细胞的收缩力和Ca2+流入影响
3 讨 论
哮喘是一种复杂的疾病; AHR是哮喘表现的核心,与ASM收缩变化密切相关[7]。Ca2+信号对ASM细胞收缩、生长和迁移至关重要。SOCE通道是Ca2+进入ASM细胞的普遍方式之一[8]。STIM1是内质网中的Ca2+传感器,其在限定的内质网/质膜连接区域寡聚化,以激活Orai1形成SOCE通道[9]。STIM1/Orai1偶联的机制包括将STIM1的约100个氨基酸的C端卷曲螺旋区与Orai1四聚体的C端和N端直接结合[10]。哮喘中的ASM收缩变化与Ca2+处理异常以及离子通道及泵的表达变化有关[11]。本研究数据表明,气道上皮源性STIM1增加可能与ASM收缩和AHR有关,表明STIM1可能是一种潜在的内皮源性收缩因子。
上皮细胞是抵抗气道中有毒和过敏物质的第一道防线。除了物理屏障功能外,气道上皮还通过释放环氧合酶产物和内皮源性收缩因子在ASM张力的调节中发挥作用。本研究表明,STIM1主要定位于支气管上皮,对过敏性哮喘小鼠模型鼻内给予sh-STIM1可降低AHR。IL-13是一种Th2型细胞因子,与过敏性哮喘的发病有关[12]。IL-13除了对杯状细胞增生和增加黏液产生作用外,还可直接调节ASM的高收缩性,从而诱导哮喘患者的AHR[13]。此外, IL-13增加了HASMC对各种收缩剂(如组胺)的Ca2+反应[14]。这些结果强调了IL-13在过敏性哮喘AHR中的作用。本研究中, STIM1敲低可降低哮喘小鼠的AHR和炎症反应,表明STIM1对哮喘具有恶化作用, STIM1的过度活跃可能是对IL-13诱导的炎症应激反应的一种代偿机制。这一发现与既往研究[15-16]一致,表明STIM1是一种应激反应蛋白,参与诱导组织损伤,如急性肺损伤和心肌梗死。
ASM的超收缩性与哮喘的AHR密切相关[7]。一般ASM收缩是在气道炎症的背景下研究。然而,在使用胶原凝胶收缩试验对单个ASM细胞进行的研究中, ASM收缩不仅与气道炎症有关,还可能是由哮喘固有异常引起的,该异常在没有哮喘气道环境的原代培养中持续存在,表明哮喘ASM发生表观遗传或信号通路重塑[17]。ASM收缩是由增加的胞浆Ca2+水平引起的,随后开始形成Ca2+-钙调蛋白-肌球蛋白轻链激酶复合物,激活Oria1介导的SOCE。在此基础上,本研究探讨了STIM1对ASM收缩的作用。本研究结果显示,鼻内滴注sh-STIM1逆转了OVA诱导的过敏小鼠AHR。sh-STIM1通过抑制钙依赖性Oria1表达,进而抑制HASMC收缩性。由于SOCE对调节HASMC的Ca2+稳态至关重要,Oria1与STIM1的相互作用对SOCE具有正性调节作用[18]。因此, STIM1通过直接调节ASM内在收缩和间接控制过敏性哮喘炎症的关键特征来促进AHR, 从而使STIM1成为内皮源性收缩因子。
综上所述,本研究证明上皮细胞中STIM1调节ASM收缩和AHR, 其作用机制与激活Oria1介导的SOCE有关。这些数据为整合哮喘上皮细胞和ASM细胞相互作用以及增强AHR的机制提供了新见解,进而促进了对哮喘发病机制的深入理解。
