高效液相色谱-串联质谱法同时测定动物源性食品中15种禁限兽药残留
2023-02-06王雪松苑中策董洁琼王圆圆郑百芹李明阳雷舒涵庞学良
李 宁,肖 琎,王雪松,苑中策,董洁琼,王圆圆,郑百芹,李明阳,雷舒涵,庞学良,吴 桐
(1.唐山市食品药品综合检验检测中心,河北唐山 063000;2.遵化市畜牧水产技术服务中心,河北遵化 064200;3.唐山市农业综合行政执法支队,河北唐山 063000)
随着我国经济迅速发展,肉类食品消费量不断增长,质量安全问题也日益凸显(刘柏林等,2021)。农业部235号公告中明确规定禁止用于所有食品动物和不得在动物性食品中检出的药物有三十余种(王鹤佳等,2021;杨丽君等,2015)。这些药物残留在动物性食品中会成严重危害人体健康(方从容等,2018)。目前,禁限兽药残留的检测方法大多是针对某种或者某类兽药,因此,建立一种同时检测多类违禁药物的方法具有一定的现实意义(程晓宏等,2022;韩瑨烜等,2022;怀文辉等,2018)。
本研究采用高效液相色谱-串联质谱法,以猪、鸡、牛、羊肉基质为样本,β-受体激动剂类、硝基咪唑类等15种禁限兽药残留为待测组分,对前处理条件、液相色谱、质谱条件进行优化,建立一种多种类禁限兽药同时检测的方法,以期为动物源性食品中禁限兽药分析和风险监测提供数据支撑(朱姗姗等,2020)。
1 材料与方法
1.1 仪器与试剂 高效液相色谱仪、5500+三重四极杆联用质谱仪(美国AB);涡旋混匀仪(IKA);氮气发生器(北京中惠普分析技术研究所);超声波清洗器(昆山市超声波仪器有限公司)。
甲硝唑等15种标准品均购自天津阿尔塔科技有限公司;甲醇、乙腈(Dikma);SCRS净化柱、萃取盐包(Cyan科技公司);五氟苯基色谱柱(美国 Phenomenex)。
1.2 标准溶液配制及标准曲线绘制 标准物质中间液(10μg/mL)配制:分别将甲硝唑等15种标准品(浓度均为100μg/mL)用甲醇稀释定容,-20℃避光保存。
混合标准物质工作液(100 ng/mL)配制:准确移取甲硝唑等15种标准物质中间液各100μL于10 mL容量瓶中,甲醇定容,4℃避光保存。
用 工 作 液 配 制 浓 度 范 围 为 0.5、2、5、20、50 ng/mL系列溶液,现配现用。
1.3 样品前处理 样品前处理:准确称取2 g均质样品于50 mL离心管1(含三颗锆珠)中;加入 2 mL纯净水,震荡涡旋 30 s;加入 1% 乙酸-乙腈溶液10 mL,震荡涡旋5 min;超声提取15 min,8000 r/min 离心 5 min,将上清液转移至50 mL离心管2中;重复提取一次,将提取液合并至离心管2中,并加入萃取盐包,立即震荡涡旋30 s,8000 r/min 离心 5 min,SCRS 净化柱净化,取 5 mL净化液氮吹至近干,用 1 mL甲醇 -水(1:1)复溶,过0.22μm微孔滤膜,供仪器分析测定。
1.4 HPLC-MS/MS条件
1.4.1 液相色谱条件 色谱柱:五氟苯基色谱柱100A(2.6μm,100×2.1 mm);柱 温 :40 ℃ ;进样体积:2μL;流速为0.4 mL/min,流动相:溶剂A为0.1%甲酸-水;溶剂B为乙腈,梯度洗 脱 程 序 :0 ~ 0.5 min,1%B ;0.5 ~ 0.6 min,10%B ;0.6 ~ 8 min,100%B ;8.0 ~ 10 min,1%B。
1.4.2 质谱条件 ESI+模式;电喷雾电压5500 V;离子源温度:500℃;气帘气:35 psi;辅助气 1:55 psi;辅助气 2:55 psi;多反应 MRM扫描。质谱参数见表1。

表1 高效液相色谱-串联质谱分析参数
2 结果与讨论
2.1 样品前处理优化 在5 ng/kg添加水平下,15种禁限兽药用乙酸乙酯、乙腈、甲醇、1%(V/V)乙酸-乙腈溶剂提取,目标组分最低回收率分别为45%、67%、58%、77%。试验发现,经涡旋震荡后再经超声助提可明显提高提取效率。与涡旋震荡10 min相比,涡旋震荡5 min加标回收率无差异。超声10、15、20 min的最低回收率分别为68%、77%、78%。若提取一次,加标回收率范围为44%~82%,重复提取为77%~104%。比较了无水NaSO4和无水MgSO4为主要成分的萃取盐包,采用 2 g无水 NaSO4和 1 g无水 MgSO4混合萃取盐包除水效果较好。还比较了Prime HLB、SCRS净化柱两种固相萃取柱的净化效果,加标回收率范围分别为55%~96%、77%~104%。
2.2 基质效应 为了降低基质效应,评价了在4种不同基质中,15种禁限兽药的响应值,以样品前处理的空白基质和50%乙腈-水分别稀释标准品配制标准曲线,以二者的斜率计算标准曲线。4种基质中15种禁限兽药的ME值在0.851~1.079之间,说明SCRS净化柱净化效果显著。
2.3 液相色谱与质谱条件的优化 为了减少上机溶剂效应的干扰,采用乙腈为有机相。比较了水、0.1%甲酸-水作为水相,表明0.1%甲酸-水作为水相各组分的响应值较好,峰形呈现较好。另比较 Waters T3 色谱柱(100 mm×2.1 mm,1.8μm)、五 氟 苯 基 色 谱 柱(100 mm×2.1 mm,2.6μm)响应值及色谱峰形,表明五氟苯基色谱柱比T3色谱柱响应值高13%。将15种禁限兽药的混合标准工作液配置成浓度为2 ng/mL,采用MRM模式,以ESI+模式优化各个离子对的去簇电压、碰撞能量等质谱参数。
2.4 线性关系及检出限、定量限 由表2可知,将15种禁限兽药标准工作液用50%乙腈-水作为溶剂,配制浓度范围为 0.5、2、5、20、50μg/L 的混合标准系列溶液,现配现用,绘制标准曲线。结果发现,15种禁限兽药在0.5~50μg/L的范围线性关系良好,相关系数均大于0.998。以3倍基线噪声确定各组分的检出限,10倍基线噪声确定各组分的定量限,最低检出限均低于0.5μg/kg,定量限均低于1.0μg/kg。表明该方法具有较好的灵敏度。
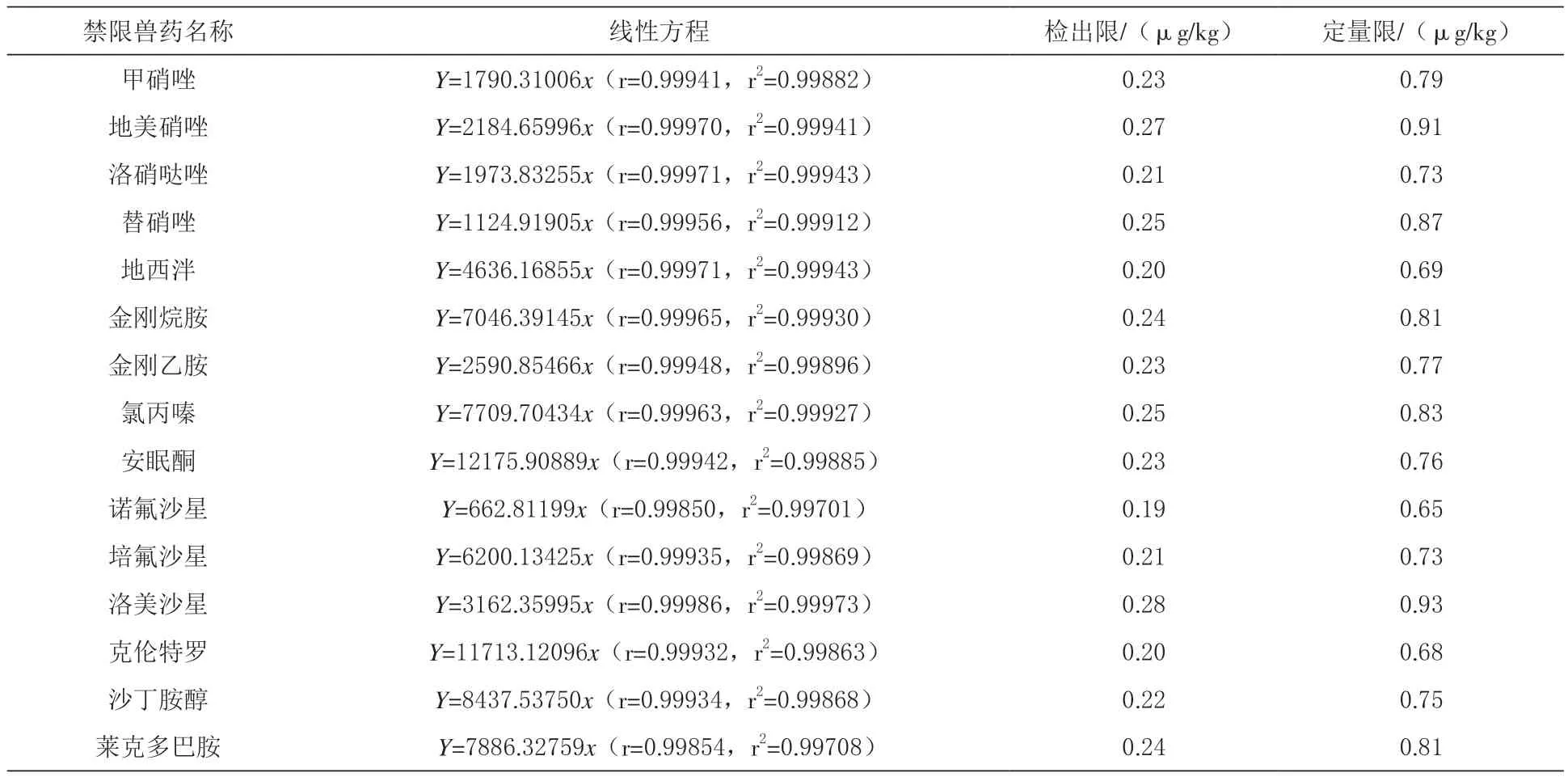
表2 15种禁限兽药的线性方程、检出限和定量限
2.5 加标回收率和精密度 由表3可知,在4种空白基质样品中添加5μg/kg的混合标准溶液进行加标回收试验,重复测定6次,结果表明,4种基质样品中15种禁限兽药的平均加标回收率为78%~98%,相对标准偏差(RSDs)为2.6%~8.3%。
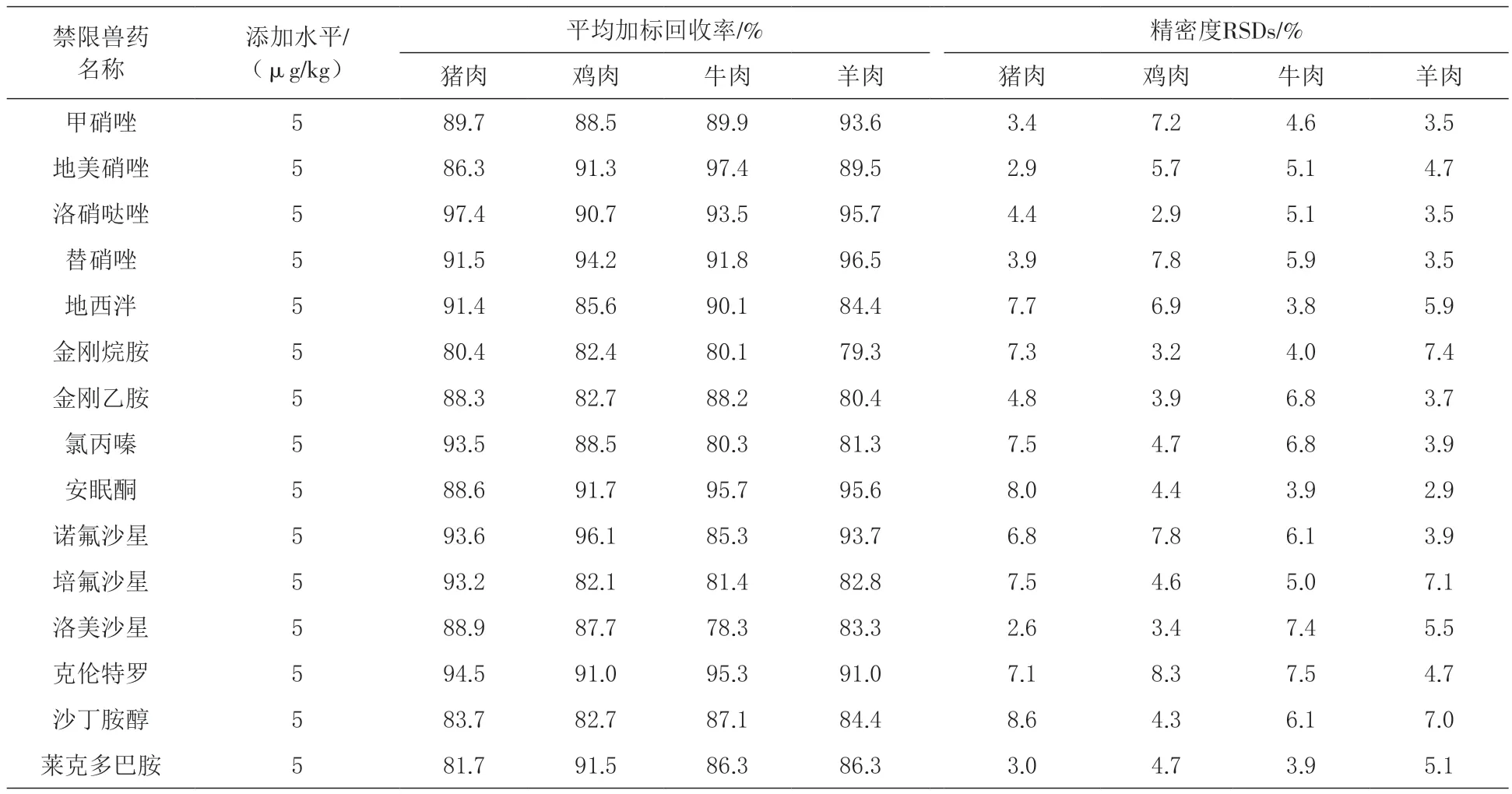
表3 猪、鸡、牛、羊肉样品中15种禁限兽药的回收率和精密度(n=6)
2.6 实际样品分析 采用该方法对随机采取的4种基质30份样品进行检测,其中鸡肉样品中检出1份甲硝唑药物残留,其定量结果为2.3μg/kg。采用国家标准(SN/T 1626-2019)进行复验,定量结果为2.0μg/kg,结果偏差为13%,说明禁限兽药在动物源性食品中依然存在添加风险。
3 结论
本文建立了4种基质中检测15种禁限兽药残留的检测方法,采用外标法定量。该方法具有操作简单、回收率高、成本低等优点,可适用于大批量样本分析,为风险监测提供有效的数据支撑。
