Recent advances in gold-complex and chiral organocatalyst cooperative catalysis for asymmetric alkyne functionalization
2023-01-30MingBaoSuZhouWenhaoHuXinfangXu
Ming Bao,Su Zhou,Wenhao Hu,Xinfang Xu
Guangdong Key Laboratory of Chiral Molecule and Drug Discovery,School of Pharmaceutical Sciences,Sun Yat-sen University,Guangzhou 5100 06,China
Keywords:Gold catalysis Chiral organocatalyst catalysis Relay catalysis Synergistic catalysis Asymmetric alkyne functionalization
ABSTRACT Homogeneous gold catalysis has demonstrated the preponderant capability of realizing a broad range of synthetically versatile alkyne functionalization over the last two decades.Though catalytic asymmetric alkyne transformation has focused on the principle of using gold catalysts either associated with chiral phosphine ligand or combined with chiral counterion,a variety of breakthroughs have been reported with the application of gold-complex and chiral organocatalyst cooperative catalysis strategy,which could enable the challenging transformations that cannot be realized by mono-catalysis with excellent stereoselectivity.This review will cover two general protocols in this field,including relay catalysis and synergistic catalysis,with emphasis on the detailed cooperative catalysts models to illustrate the roles of the two catalysts and highlight the potential synthetic opportunities offered by asymmetric cooperative catalysis.
1.Introduction
Homogeneous gold catalysis has demonstrated the preponderant capability of enabling a variety of synthetically useful alkyne transformations over the last two decades[1–9].Together with the development of versatile phosphine ligands and gold-complexes,this area has attracted intensive attraction and led to immense review articles on catalytic alkyne functionalization that are classified according to the different types of reactions[10–15],catalysts or catalytic methods[16–21]and synthetic applications[22–24].However,the discovery of the catalytic asymmetric versions of gold-catalyzed alkyne functionalization is relatively slow,which mainly relies on the using of chiral gold-complexes,either associated with chiral phosphine ligand or combined with chiral counterion,for the asymmetric induction[20–21].
Since the first example of asymmetric gold-catalyzed aldol reaction,which was reported by Ito,Sawamura,and Hayashi in 1986[25],the chirality induction that controlled by a chiral ligand coordinated to the metal catalyst has been considered as the general principle in asymmetric gold catalysis[26–28].Whereas,only a handful of chiral gold-complexes have been disclosed to achieve high enantioselectivity in catalytic alkyne functionalization[29,30].This is mainly ascribed to the linear geometry of gold-alkyne coordinated complex,in which,the chiral ligand located far away from the reaction center,and the addition of the nucleophile to the activatedπ-bond occursviaan outer-sphere mechanism(Scheme 1,model a).Thus,it is an appealing and persistent research focus for realizing the asymmetric alkyne functionalization with the development of effective and robust gold catalysts and catalytic methods.
The second principle that has been applied to the homogeneous gold catalysis is asymmetric counteranion-directed catalysis(Scheme 1,model b,ACDC)[31,32].In 2007,Toste’s group has reported a chiral gold phosphate catalyst,which generated from LAuCl complex and chiral binaphthol-derived silver phosphate(Scheme 1,Eq.a),promoting the hydroalkoxylation of allenols with high enantioselectivity[33,34].Soon after this landmark report,a variety of catalytic asymmetric alkyne transformations with analogous ACDC strategy(Scheme 1,Eqs.a and b)have been disclosed by Dixon[35],Gong[36,37],and many others[38–43]independently.In comparison to Toste’s work,these later advances feature a binary catalytic system,in which the chiral Brønsted acid could be used in an excess amount related to the gold-complex,enabling one-pot sequential/cascade reactions through a cooperative catalysis strategy[44–46].Moreover,by incorporation of phosphine ligand with chiral Bronsted acid,which was termed as tethered counterion-directed catalysis(TCDC),has been proposed by Marinetti and Guinchard as a complement to ACDC strategy in asymmetric alkyne transformations[47,48].
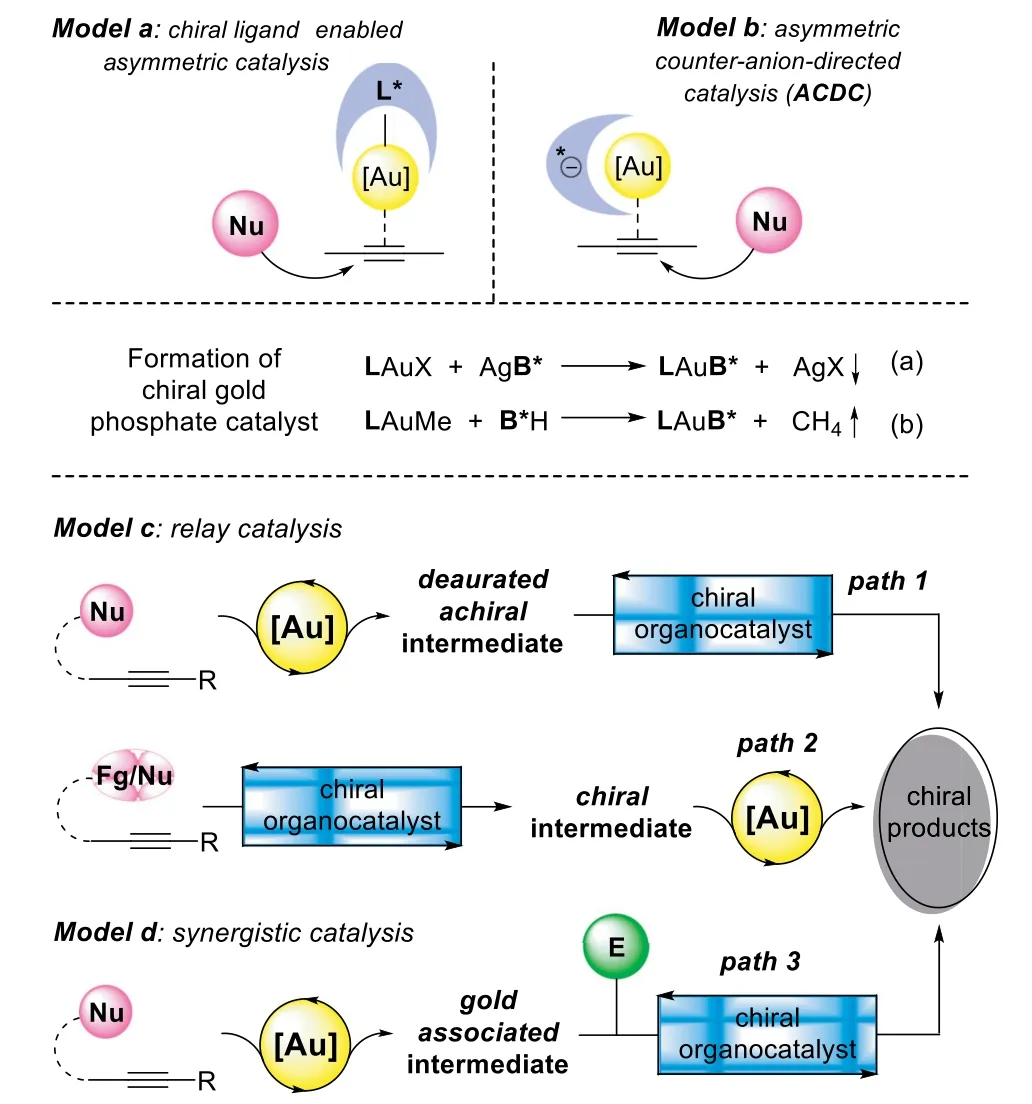
Scheme 1.Catalytic strategies for asymmetric alkyne functionalization.
Fig.1.Structures of ligands in generally used gold-complexes.
In contrast to the mono-catalysis,the cooperative catalysis has shown several benefits[49–55],especially in asymmetric goldcatalyzed alkyne functionalization reactions,which can 1)avoid the tedious preparation of complex chiral gold catalysts through a flexible combination of the gold catalyst with compatible chiral cocatalyst;2)improve the efficiency or enabling previously unattainable alkyne transformation by activation both of the assembling species;3)achieve high stereoselectivityviaselective activation of the prochiral substrate with a chiral cocatalyst,which is much closer to the addition center in comparison to the ligand on the gold-complex.
Early reviews related to the topic of gold and chiral organocatalyst cooperative catalysis have been published by Hashimi[44],Enders[45]and Patil[46]in 2010,2012 and 2014,respectively,in which the organocatalysts limited to the chiral binaphthol-derived phosphoric acids(CPA).Recently,the metal-catalyzed asymmetric sequential hydrofunctionalization of alkynes has been reviewed by Lu[56].However,no critical review that summarized the catalytic asymmetric alkyne functionalization with gold-organo cooperative catalysis strategy has been reported so far,although some of these advances have been selectively highlighted in a few of review articles on the theme of metal-organo binary catalysis[49–53].
In the past decade,the enantioselective alkyne functionalization reactions have sharply increased by taking advantage of goldcomplex(Fig.1)and chiral Brønsted acid(Fig.2)cooperative catalysis.A variety of asymmetric cascade transformations have been disclosed through different catalytic models,including relay catalysis(Scheme 1,model c)and synergistic catalysis(Scheme 1,model d).Moreover,in addition to chiral Brønsted acids,a number of chiral organocatalysts have been introduced as gold compatible cocatalysts,such as chiral amine catalysts,urea catalysts,cinchona alkaloids,and squaramide-type catalysts.Thus,a critical review paper highlighting the recent advancements in gold and chiral organocatalyst cooperative catalyzed asymmetric alkyne functionalization is timely and desirable.
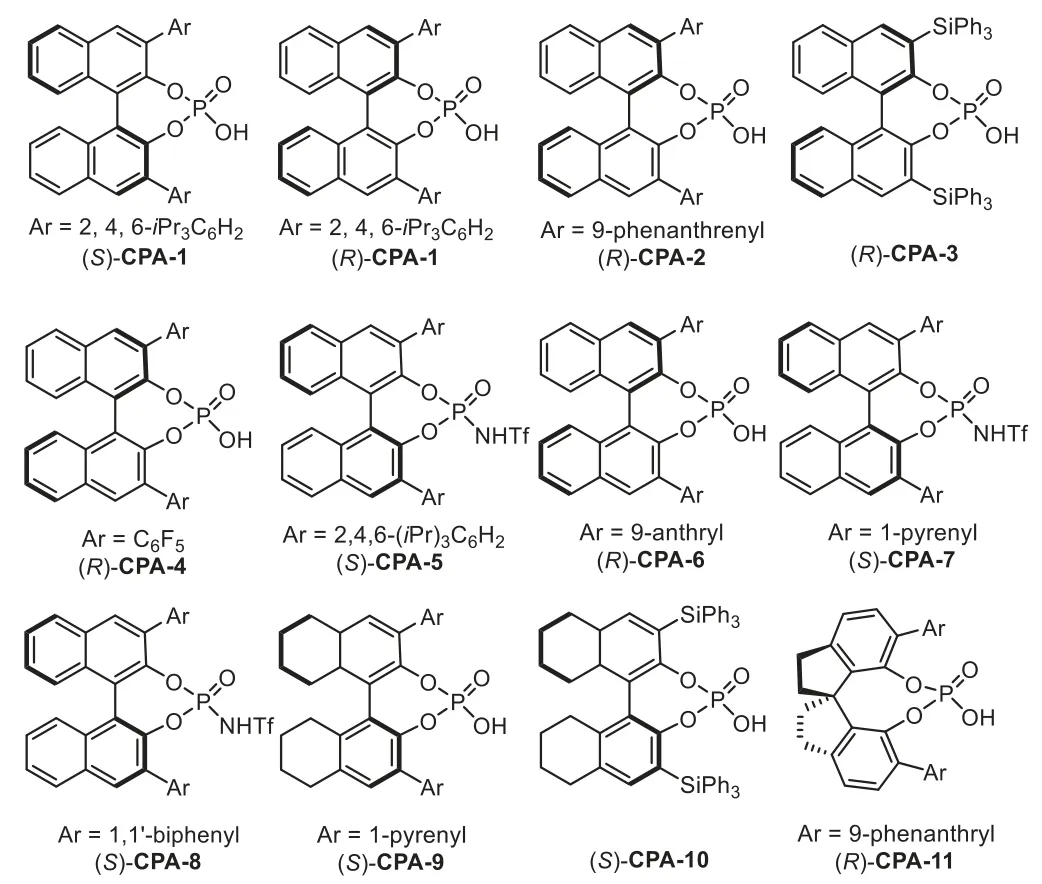
Fig.2.Structures of generally used chiral Brønsted acids.
Scheme 2.General sequence of gold-complex and chiral Brønsted acid relay catalysis.
This review will cover two protocols in this field,including relay catalysis and synergistic catalysis(Scheme 1,models c and d),with emphasis on the detailed cooperative catalysis patterns for the asymmetric induction to illustrated the roles of the two catalysts,and highlight the potential synthetic opportunities offered by these robust and efficient cooperative asymmetric catalysis methods.On the other hand,the gold and chiral Lewis acid cooperative catalysis has also been proved to be a practical protocol for the asymmetric alkyne functionalization.Representative advances in this area have been reported independently by Hashmi[57],Feng[58–61]and few others[62,63].Due to space limitation,these advances will not be included in this review article.
2.Gold-complex and chiral Brønsted acid relay catalysis
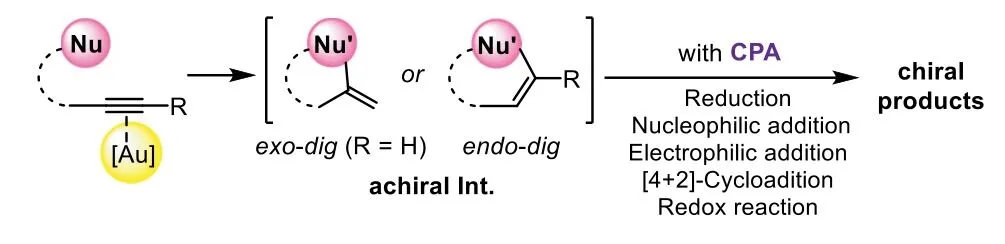
The general model of gold-complex and chiral Brønsted acid relay catalysis follows an order of gold-catalyzed hydrofunctionalizationvia exo-digorendo-digcyclization to form an achiral enol/enamine intermediate,which could be terminated by different asymmetric transformations in the presence of corresponding chiral phosphoric acids(CPA),including asymmetric reduction,nucleophilic addition,electrophilic addition,[4+2]-cycloaddition,and redox transformation(Scheme 2).
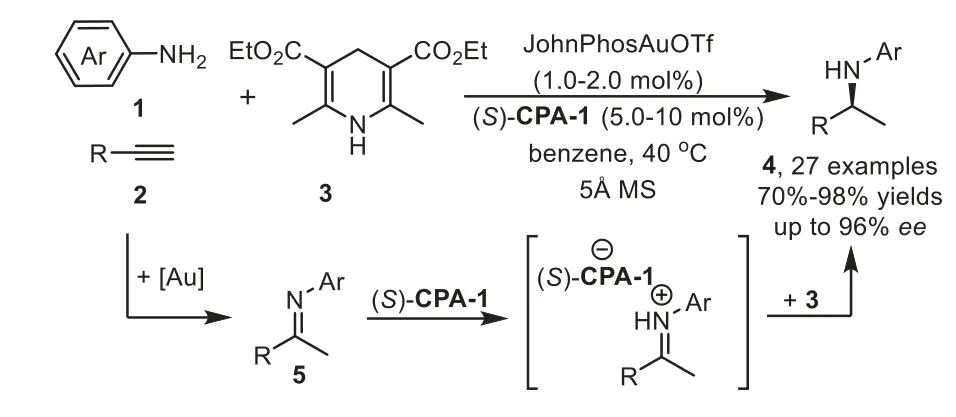
Scheme 3.Gold-complex and chiral phosphoric acid relay catalysis for the synthesis of chiral secondary amines.
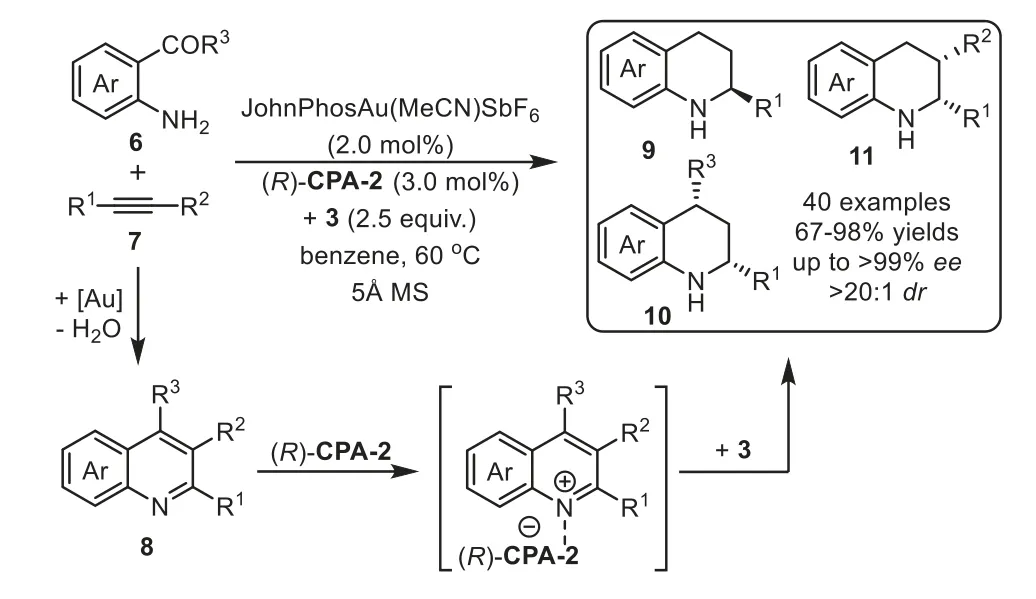
Scheme 4.Gold-complex and chiral phosphoric acid relay catalysis for the synthesis of chiral tetrahydroquinolines.
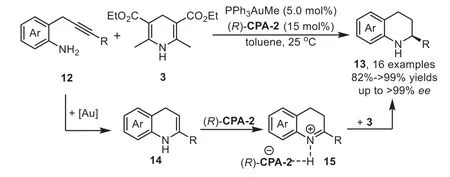
Scheme 5.Gold-complex and chiral phosphoric acid relay catalysis for the synthesis of 2-substituted tetrahydroquinolines.
Firstly,the catalytic hydroamination could be terminated by asymmetric transfer hydrogenation/reduction reaction.In 2009,Che and coworkers reported a highly enantioselective tandem intermolecular hydroamination/asymmetric transfer hydrogenation cascade reaction of anilines 1 with terminal alkynes 2[64].The proposed reaction mechanism involves gold-catalyst promoted intermolecular hydroamination of alkyne with amine to generate imine species 5,followed by enantioselective transfer hydrogenation with Hantzsch ester 3 in the presence of chiral phosphoric acid(S)-CPA-1viaforming iminium salt,affording chiral secondary amine 4 in good to high yields with generally excellent enantioselectivity(Scheme 3).Later in 2012,the same group developed an efficient one-pot method for the asymmetric synthesis of tetrahydroquinolines,including 2-substituted,2,3-and 2,4-disubstituted optically active tetrahydroquinolines,from 2-aminobenzaldehydes or 2-aminophenones 6 with alkynes 7viathe analogous cooperative catalytic system[65].This cascade reaction proposed to start with gold-catalyzed hydroamination-condensation to furnish a quinoline intermediate 8,which undergoes asymmetric transfer hydrogenation with a Hantzsch ester 3 in the presence of chiral Brønsted acid(R)-CPA-2 to give chiral tetrahydroquinolines 9–11 with high regio-,diastereo-and enantioselectivities(Scheme 4).
At the same time,the intramolecular version of above cascade reaction has been reported by Gong’s group in 2009[66].This reaction involves a sequence of gold-catalyzed intramolecular hydroamination of 2-(2-propynyl)aniline 12 to furnish 1,4-dihydroquinoline 14,followed by isomerization of 14 into 3,4-dihydroquinolinium with CPA and,finally,generating the optically active tetrahydroquinolines 13viaasymmetric transfer hydrogenation with Hantzsch ester 3(Scheme 5).The key to realization of this reaction would be the identification of a gold-complex that would afford an efficient hydroamination but would not affect the following enantioselective transfer hydrogenation.In this work,the chiral phosphoric acid(R)-CPA-2 was used in triple times in compared to the gold catalyst PPh3AuMe,which means the gold phosphorate might be formed in this case(Scheme 1,Eq.b).However,the control experiment with gold phosphorate was incomplete,indicating that it is the phosphoric acid which really controlled the stereochemistry.
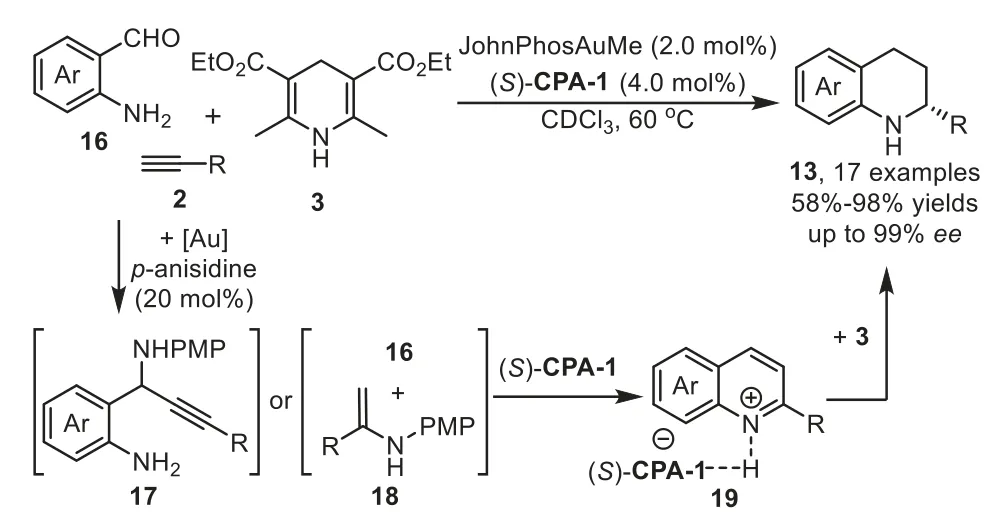
Scheme 6.Gold-complex/p-anisidine/chiral phosphoric acid relay catalysis for the synthesis of 2-substituted tetrahydroquinolines.
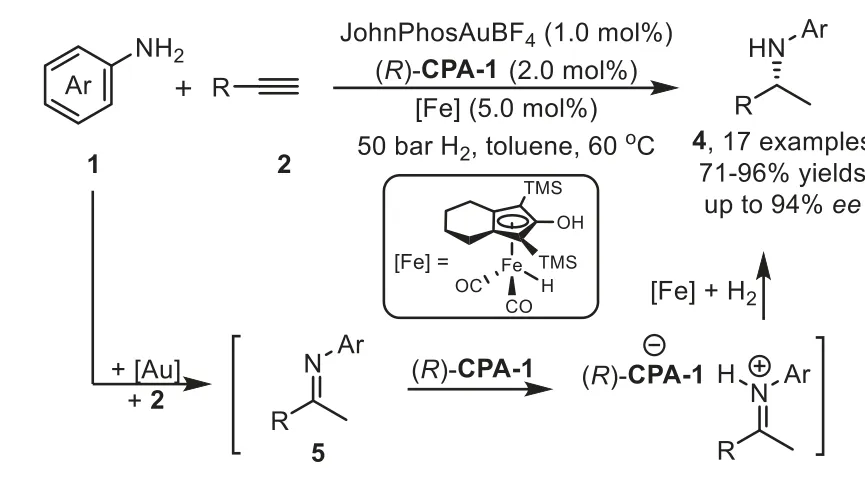
Scheme 7.Gold-complex,chiral phosphoric acid,and iron-complex relay catalysis for the synthesis of chiral amines.
In 2013,Patil reported an enantioselective cooperative triple catalysis system consisting of gold-complex/p-anisidine/chiral phosphoric acid catalysts for the synthesis of 2-substituted tetrahydroquinolines 13 from 2-aminobenzaldehydes 16,terminal alkynes 2 and Hantzsch ester 3 in good yields with excellent enantioselectivity[67].Several controlled experiments have been performed to understand the role of each catalyst,which indicate that all three catalysts,gold phosphate(generatedin situfrom gold-complex and chiral phosphoric acid),p-anisidine,chiral phosphoric acid(CPA),are necessary to obtain the final products,while the chiral phosphoric acid(S)-CPA-1 plays the key role to promote the enantioselective transfer hydrogenation of quinolinium 19,which generatedin situfrom 17 or 18,with Hantzsch ester 3 to form chiral 2-substituted tetrahydroquinolines 13 in good to high yields(Scheme 6).
In addition to the transfer hydrogenation with Hantzsch ester,the catalytic asymmetric reduction ofin situgenerated imine intermediate is an atom-economic approach for the synthesis of chiral amines.In 2012,Beller and coworkers demonstrated an enantioselective reductive hydroamination of alkynes 2 with anilines 1 for synthesis of highly enantiomerically enriched chiral secondary amines 4 by utilizing a gold-complex,Knölker’s iron complex,and chiral phosphoric acid ternary catalytic system[68].Different from the Che’s work[64],the iminium ion intermediate in this reaction is reduced directly by Knölker’s iron complex in the presence of hydrogen in one-pot,instead of using Hantzsch ester 3(Scheme 7).
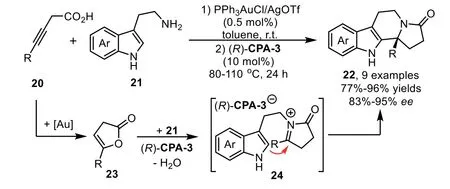
Scheme 8.Gold-complex and chiral phosphoric acid relay catalysis for the synthesis of tetracyclic indole derivatives.
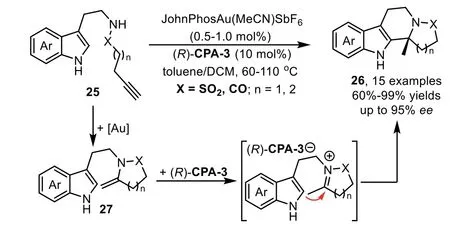
Scheme 9.Gold-complex and chiral phosphoric acid relay catalysis for the synthesis of tetracyclic sulfonamide scaffolds.
Catalytic hydrofunctionalization could also be terminated by asymmetric nucleophilic addition reaction.Generally,the enamine or enol intermediates,which generatedviagold-catalyzed hydrofunctionalization of alkynes,could be converted to corresponding electrophilic iminium/oxonium speciesin situin the presence of Brønsted acid.Thus,a variety of nucleophiles,such as indoles,electronic-rich arenes and alcohols could be used to intercept the iminium/oxonium speciesvianucleophilic addition reaction.In 2009,Dixon reported a gold and chiral phosphoric acid relay-catalyzed enantioselective cascade reaction of alkynoic acids 20 with tryptamines 21,which led to an efficient synthesis of enantiopure tetracyclic compounds 22 in good yields with high to excellentee[69].The reaction is initiated with gold-catalyzed 5-endo-digcyclization of 20 to form five membered enol lactone 23.In the presence of chiral phosphoric acid,(R)-CPA-3,the enol lactone is attacked by the amine moiety of 21 to generate ketoamide 22,which undergoes a dehydrative cyclization throughNacyliminium intermediates 24.The presence of the chiral counter anion allows successful stereocontrol in the nucleophilic addition step with tethered indole,leading to the polycyclic indole derivatives in good to excellent yields and enantioselectivity(Scheme 8).Moreover,the low gold catalyst loading is essential for obtaining the products in highee,because an excess amount of PPh3AuCl/AgOTf might generate residual TfOH,which could lead to cyclization products with poor enantioselectivity.
In 2013,a highly enantioselective intramolecular hydroamination/cyclization cascade reaction has been reported by the Dixon’s group for the synthesis of polycyclic sulphonamide scaffolds 26 from functionalized alkynes 25 under the relay catalysis of goldcomplex and chiral phosphoric acid in excellent yields and stereoselectivity[70].Mechanistic studies reveal that the reaction proceeds through a gold-catalyzed intramolecular hydroamination of alkynyl sulphonamide 25 to obtain the five membered cyclic enamine intermediate 27,followed by an enantioselective cyclization through iminium intermediate with tethered indole to form the indole derivatives(Scheme 9).High enantioselectivity is expected when the cyclization is triggered by chiral phosphoric acid(R)-CPA-3viaforming chiral ion pair,instead of background reaction promoted by gold-catalyzed cyclization.
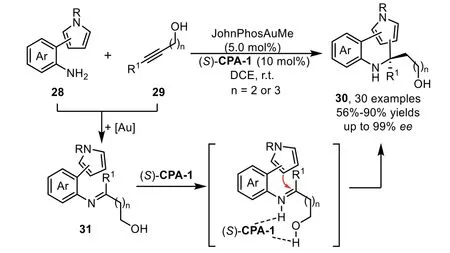
Scheme 10.Gold-complex and chiral phosphoric acid relay catalysis for the synthesis of pyrrole-containing N-heterocycles.

Scheme 11.Gold-complex and chiral phosphoric acid relay catalysis for the synthesis of spiroacetals.
Two years later,a two-component enantioselective hydroamination/cyclization cascade reaction has been reported by the Patil’s group with pyrrole tethered aromatic amines 28 and alkynes 29 under the relay catalysis of gold-complex and chiral phosphoric acid[71].In this reaction,a variety of pyrrole-based aromatic amines,including 1-,2-,or 3-subsitutued pyrroles,could be well tolerated,providing pyrrole-embeddedN-heterocycles bearing a quaternary carbon center with structural diversity(Scheme 10).In addition,from the control experiments and DFT calculations,the tethered hydroxyl species in the alkyne part turns out to be crucial for obtaining products in high yields and enantioselectivities,which might interact with the chiral phosphoric acid through hydrogen-bonding.
In the same year,Rexit’s group disclosed an asymmetric cascade reaction for the synthesis of optically active spiroacetals 33 in excellent yields and high enantioselectivities under the relay catalysis of gold-complex and chiral phosphoric acid[72].In this reaction,the gold-catalystin situgenerated from a methyl gold-complex and phosphoric acid promotes the initial cyclization reaction of alkynyl glycols 32,while the chiral phosphoric acid(S)-CPA-1 dominantly catalyzes the following spiroacetalization and controls the stereoselectivity(Scheme 11).Moreover,this protocol provides an efficient alternative strategy for the synthesis of enantioenriched spiroacetals by essentially avoiding the preparation of cyclic enol ethers.
At the same time,Jia and co-workers successfully developed a novel and efficient redox annulation reaction of nitroalkynes 36 with indoles 37 by a gold(III)/chiral phosphoric acid(CPA)dual catalysis,which provides a facile access to indolin-3-ones 38 bearing C2 quaternary stereocenter in high yields and excellent enantioselectivities[73].The reaction mechanism is that the nitro group attacked the gold-activated alkyne speciesviaa 5-exo-digcyclization to formα-oxo gold carbene intermediate,followed by an intramolecular trapping with the nitroso group to generate the isatogen compound 39.Subsequently,the chiral ion pair 40 was formedin situfrom 39 in the presence of(R)-CPA-4,followed by nucleophilic addition with indole 37 to produce indolin-3-one derivatives 38(Scheme 12).
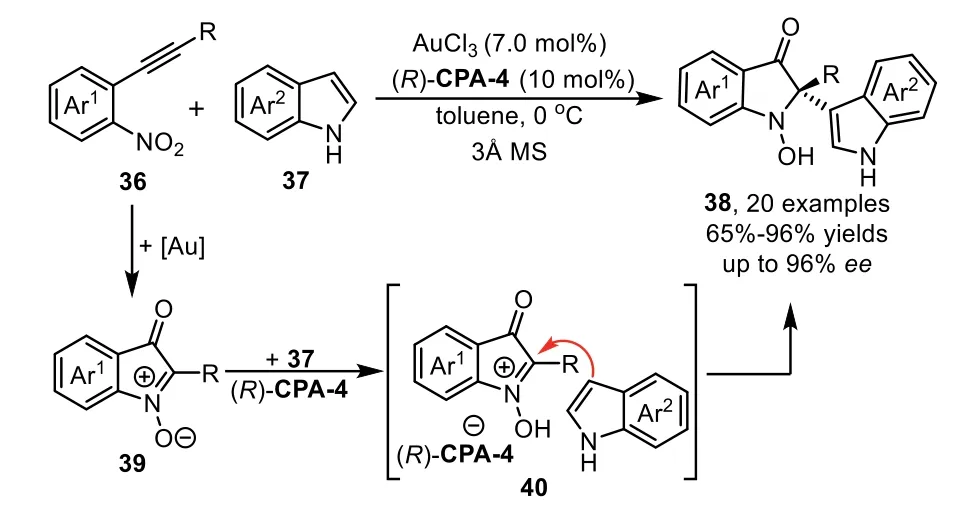
Scheme 12.Gold-complex and chiral phosphoric acid relay catalysis for the synthesis of indolin-3-ones.
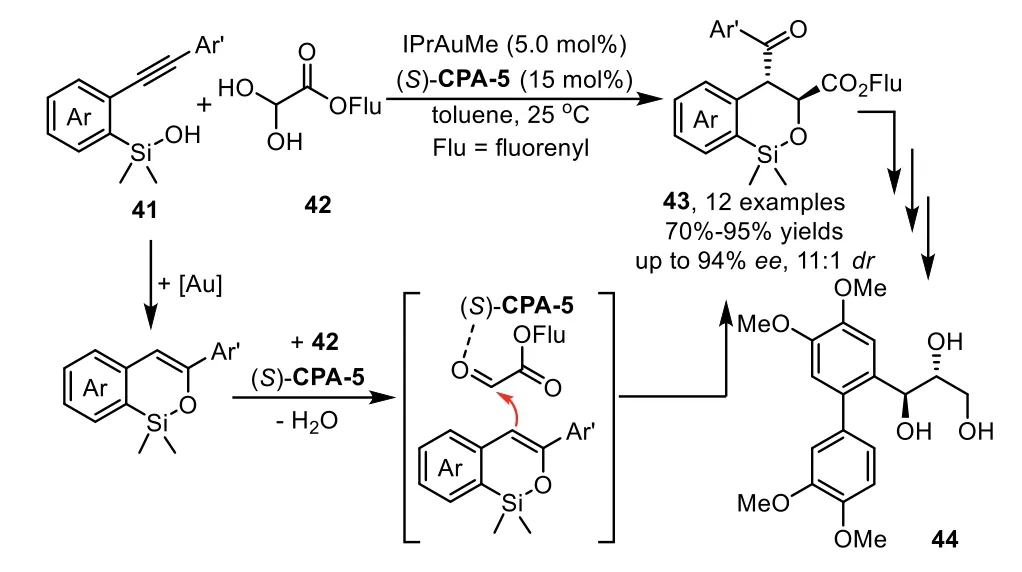
Scheme 13.Gold-complex and chiral phosphoric acid relay catalysis for hydrosiloxylation/Mukaiyama Aldol cascade reaction.
In addition to the cascade reaction that terminated by asymmetric nucleophilic addition,thein situgenerated enamine or enol intermediates could be directly intercepted by electrophilesviaenantioselective Aldol and Michael additions.In these cases,the chiral phosphoric acids(CPA),instead of interaction with nucleophiles,activate the electrophiles through hydrogen-bonding with the carbonyl oxygen.In 2013,Gong and co-workers reported a practical cascade hydrosiloxylation/Mukaiyama Aldol addition reaction of arylacetylenes 41 with glyoxylates 42 in high yields with excellent enantioselectivities by using an IPrAuMe and chiral Brønsted acid(S)-CPA-5 binary relay catalysis system[74].During the condition optimization studies,they found that the use ofN-heterocyclic carbene(NHC)IPr as ligand turned out to be crucial for obtaining the cyclic products 43 with good yields andee(Scheme 13).Notably,higher enantioselectivity is observed when steric bulky substrate,such as fluorenyl(Flu)glyoxylates,were used as acceptors for the addition with enol intermediate.In this work,the siloxy group is an attractive species for a variety of transformations.For instance,the cyclic product could be converted to an optically active triol 44 in three steps,which could serve as a potential intermediate for the synthesis of metasequirin B.Moreover,this method could be readily scaled up to a 10-g scale with lower catalyst loading,which shows potential application to organic synthesis.In the same year,Wu and coworkers have established an enantioselective approach with above asymmetric cascade reaction as the key step for the synthesis of(−)-5–epi-eupomatilone-6.The total synthesis involves a sequence of intramolecular hydrosiloxylation,asymmetric Mukaiyama aldol reaction,Arndt-Eistert homologation,and Suzuki coupling reaction,providing the final product in about 9%overall yield(Scheme 14)[75].
In 2016,Han and co-workers disclosed an intramolecular hydroamination/Michael addition cascade reaction enabled by goldcomplex/chiral phosphoric acid(CPA)relay catalysis,leading to chiral tetrahydrocarbazoles(THCs)46 bearing two adjacent stereocenters with good to excellent yields and high enantioselectivities(Scheme 15)[76].The control experiment indicates the presence of intermediate 47,although it is too reactive to isolate,this intermediate could direct lead to the racemic Michael addition product 46 without the assistance of CPA,which might be the main reason for the observed moderate enantioselectivity in some cases.
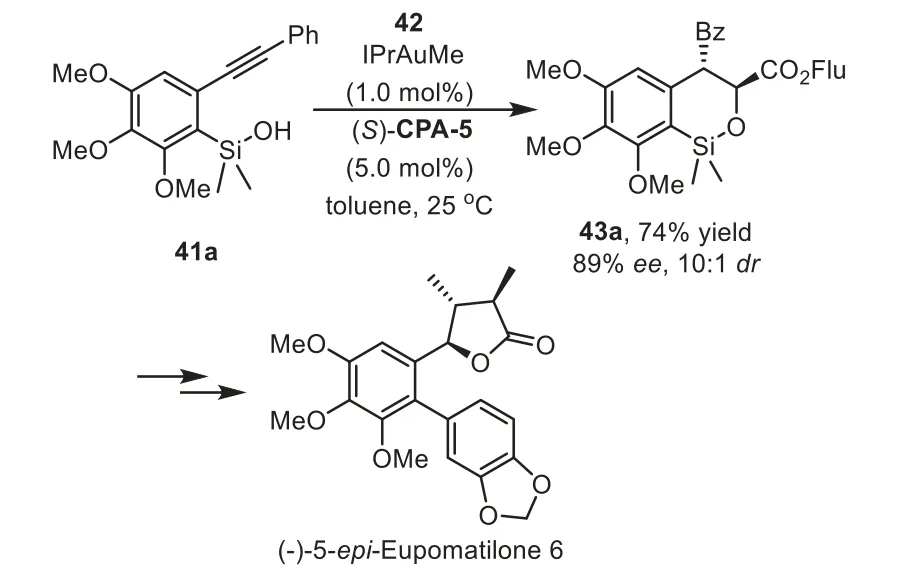
Scheme 14.Application to the enantioselective hydrosiloxylation/Mukaiyama aldol cascade reaction for the synthesis of(−)−5–epi-eupomatilone-6.
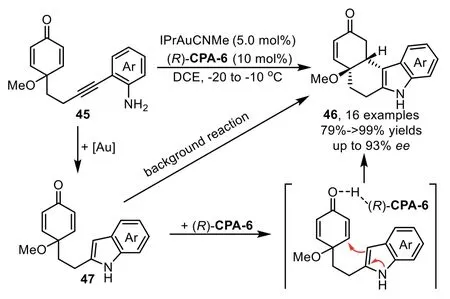
Scheme 15.Gold-complex and chiral phosphoric acid relay catalysis for the synthesis of tetrahydrocarbazoles.
Furthermore,the enol intermediatesin situgenerated from gold-catalyzed intramolecular hydroalkoxylation of alkynes could serve as 4-C/2-C synthons in[4+2]-cycloaddition depending on the embodied functional group in these used alkyne materials.In 2012,Gong and co-workers reported the first asymmetric hydrosiloxylation/Diels-Alder cascade reaction of enynyl silanols 48 with naphthoquinone 49 under the gold-complex/chiral Brønsted acid binary catalysis[77].The reaction proceeds through goldcatalyzed intramolecular hydrosilylation of enynyl silanol 48 to form an active silyloxydiene intermediate 51,which participates in a chiral phosphoric acid catalyzed asymmetric Diels-Alder reaction with an electron deficient olefin 49 to afford highly enantioenriched polycyclic products 50(Scheme 16).Notably,the Diels-Alder reaction between silyloxydiene species 51 and 49 do not occur in the presence of gold-complex alone,which indicates that the[4+2]-cycloaddition reaction is solely accelerated by chiral Brønsted acid(S)-CPA-7.
One year later,the same group disclosed an asymmetric multicomponent reaction of alkynols 52 with anilines 1 and salicylaldehydes 53 by using gold-complex/chiral phosphoric acid relay catalysis to give aromatic spiroacetals 54 in high yields and stereoselectivities[78].The reaction proceeds through a gold-catalyzed intramolecular hydroalkoxylation of alkynol to afford exocyclic enol ether 55,followed by an asymmetric Mannich-type reaction with salicylaldehydimines 56 that generatedin situfrom the condensation reaction of salicylaldehydes 53 and anilines 1 in the presence of chiral phosphoric acid(R)-CPA-3,producing oxonium ion 57 which is terminated by acetalization to deliver the spiroacetal products 54(Scheme 17).The overall cascade process features a[4+2]-cycloaddition by usingin situformed enol ether 55 as a 2-C synthon.The control experiment shows that both gold phosphate and chiral Brønsted acid can catalyze the cascade reaction,while(R)-CPA-3 plays a dominant role in the control of enantioselectivity in the Mannich-type addition step.
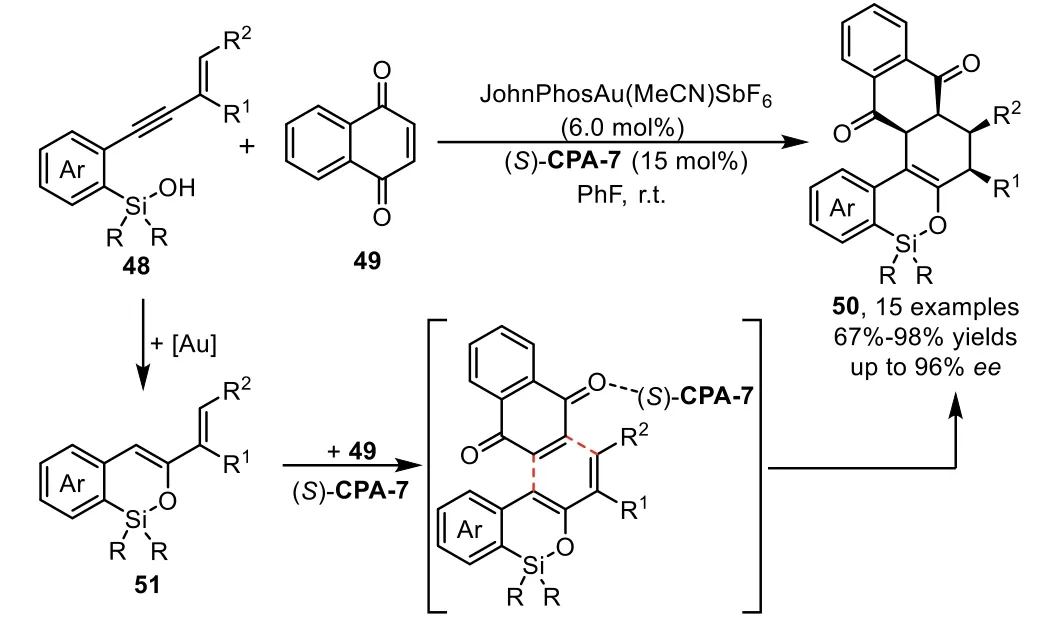
Scheme 16.Gold-complex and chiral phosphoric acid relay catalysis for the synthesis of polycyclic heterocycles.
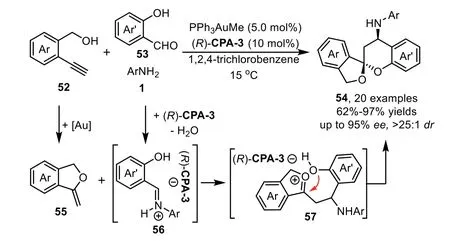
Scheme 17.Gold-complex and chiral phosphoric acid relay catalysis for aromatic spiroacetals.
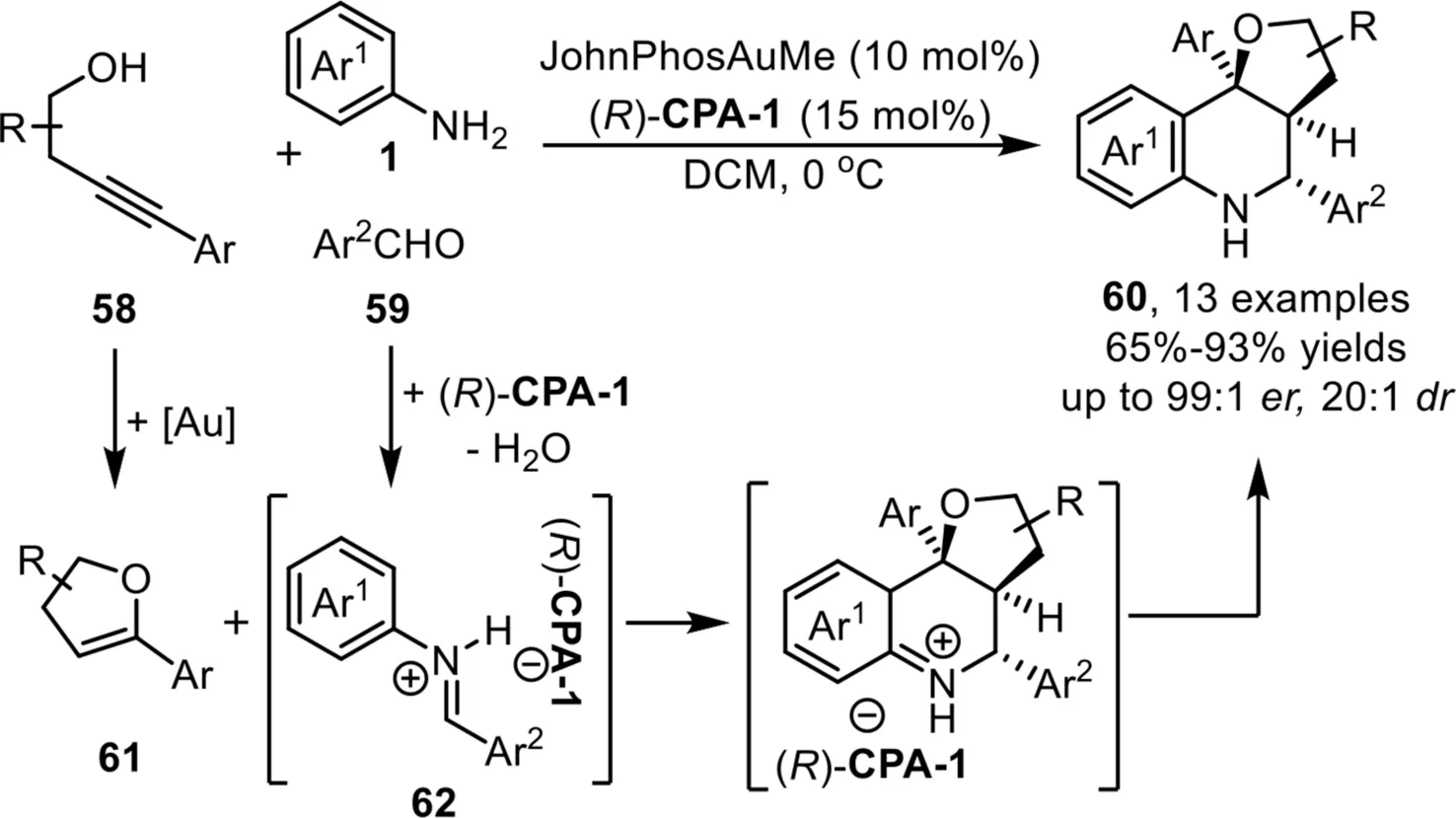
Scheme 18.Gold-complex and chiral phosphoric acid relay catalysis for the synthesis of hexahydrofuro[3,2-c]quinolines.
One year later,Rodríguez and co-workers reported an asymmetric three component coupling reaction between alkynols 58,aldehydes 59 and aryl amines 1 for the diastereo-and enantioselective synthesis of hexahydrofuro-[3,2-c]quinolines 60 under the gold-complex/chiral phosphoric acid relay catalysis system[79].Overall,the role of gold phosphate that generatedin situfrom gold-complex and chiral phosphoric acid,is to catalyze the hydroalkoxylation reaction to form enol ether 61,which serves as a 2-C synthon in the following[4+2]-cycloaddition reaction;while the function of chiral phosphoric acid(R)-CPA-1 is to promote the imine formation and control the enantioselectivity in the final Povarov reaction(Scheme 18).
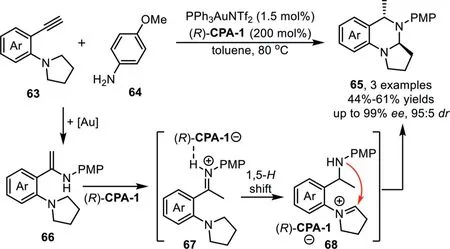
Scheme 19.Gold-complex and chiral phosphoric acid relay catalysis for the synthesis of cyclic aminals.
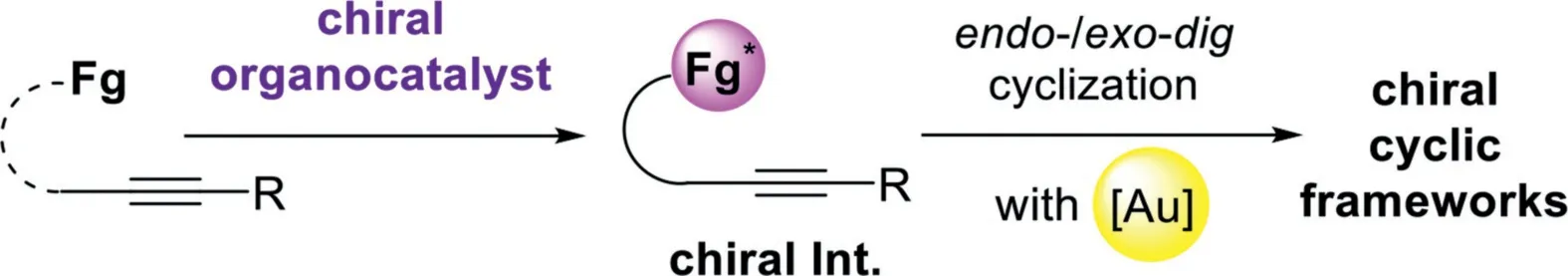
Scheme 20.Chiral organocatalyst and gold-complex relay catalysis.
The catalytic hydroamination could be terminated by redox cascade reaction.In 2013,Gong’s group reported an asymmetric relay catalytic system for the synthesis of cyclic aminals 65 from 2-ethynylanilines 63 and amines 64viaa cascade hydroamination/redox cascade reaction[80].A proposed reaction mechanism involves gold-catalyzed hydroamination of terminal alkynes withp-anisidine to form enamine intermediate 66,which could be converted to an iminium species 68 in the presence of(R)-CPA-1 through a 1,5-hydride transfer processvia67,followed by intramolecular cyclization to give cyclic aminals 65(Scheme 19)[81].In this reaction,an enhancement in enantioselectivity was observed when chiral phosphoric acid was used in excess amount,which might be due to the fact that the gold-catalyzed nonselective racemic cyclization would compete with the chiral phosphoric acid catalyzed process.
3.Chiral organocatalyst and gold-complex relay catalysis
The general catalytic model of the chiral organocatalyst and gold-complex relay catalyst follows a sequence of initially formation of chiral intermediates in the presence of chiral organocatalyst,including asymmetric Mannich-type addition,Aldol addition,[4+2]-cycloaddition,and Nazarov-type electrocyclization,then,these chiral species were terminated by a gold catalyzed intramolecular cyclization,leading to the chiral cyclic frameworks(Scheme 20).In this area,including the chiral Brønsted acid(Fig.1),a variety of other chiral organocatalysts(Fig.3)have been found to be compatible with the gold-complex to promote the asymmetric alkyne transformationsviarelay catalyst.
Chiral organocatalysts promoted asymmetric Mannich type addition is one of the general methods for the preparation of alkyne embodied chiral amine species,which could be subjected to the gold-promoted hydroamination,leading to a variety of chiralNheterocycles[82–86].In 2010,Gong’s group described the first example of gold-complex and chiral phosphoric acid catalyzed enantioselective three component cascade reaction between 2-(2-propynyl)anilines 69,aldehydes 59 and enamide 70 for the directly assembly of julolidine derivatives 71 in good to high yields[87].Mechanistically,the chiral phosphoric acid(R)-CPA-6 catalyzes formal[4+2]-cycloaddition reaction of enamide 70 with iminium species 72,which derived from condensation of 69 with 59,generating enantiopure aminoalkyne 73.Subsequently,this intermediate undergoes intramolecular hydromination catalyzed by the gold phosphate,and the stable julolidine derivatives 71 are isolated after reduction(Scheme 21).Kinetic studies reveal that the chiral phosphoric acid not only enables the asymmetric[4+2]-cycloaddition reaction,but also involves the formation of gold phosphate which catalyzed hydroamination reaction.
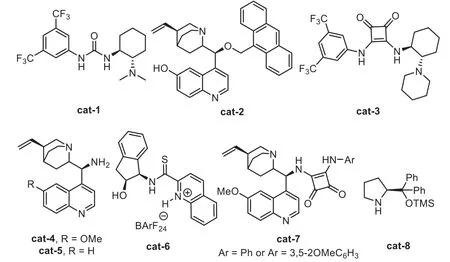
Fig.3.Structures of other gold compatible chiral organocatalysts.
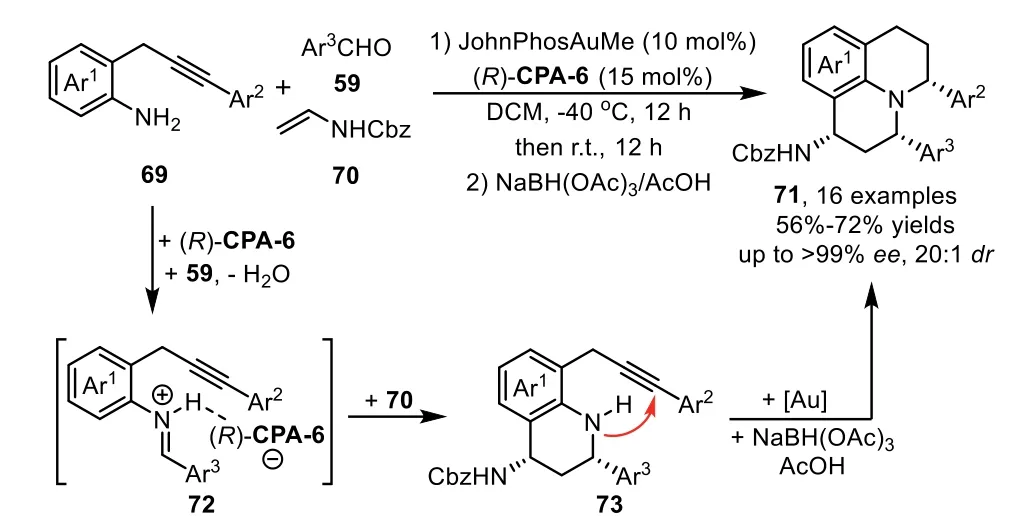
Scheme 21.Chiral phosphoric acid and gold-complex relay catalysis for the synthesis of julolidine derivatives.
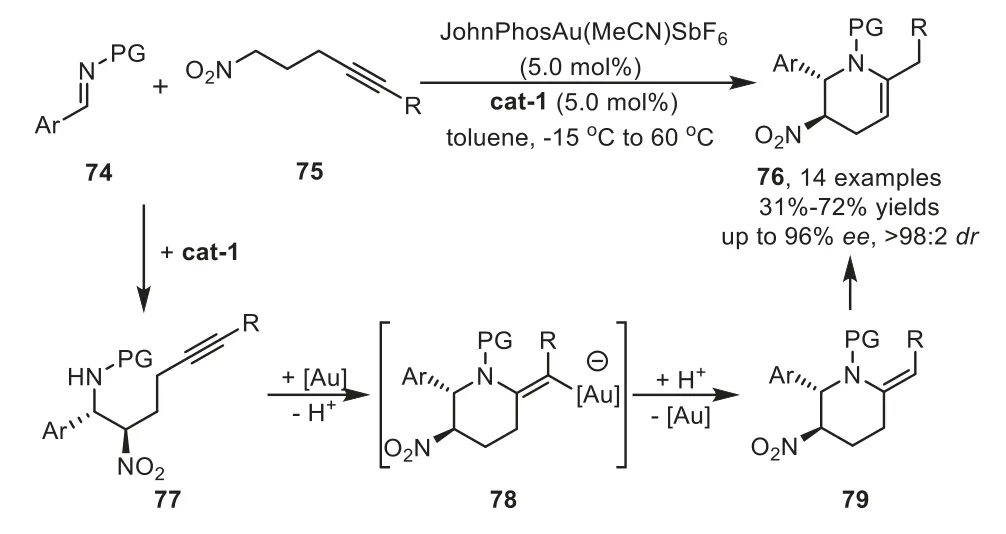
Scheme 22.Chiral urea derived organocatalyst and gold-complex relay catalysis for the synthesis of tetrahydropyridine derivatives.
Two years later,a highly enantioselective cascade reaction for the preparation of synthetically useful tetrahydropyridine derivatives 76 has been was reported by Dixon and co-workers by employing an organocatalyst and gold-complex relay catalysis[88].A proposed mechanism involves an enantioselective nitro-Mannich reaction of aldimine 74 with nitro alkyne 75 under the control of Takemoto’s chiral urea catalyst cat-1.The resultingβ-nitroamine 77 would then be subjected to cyclizeviaa hydroamination reaction using a gold catalyst to furnish piperidine intermediate 79 after protodemetalation.Subsequently,isomerization of the alkene part would lead to tetrahydropyridine products 76(Scheme 22).
Scheme 23.Chiral quinidine derived organocatalyst and gold-complex relay catalysis for the synthesis of spirooxindole derivatives.
Scheme 24.Chiral urea derived organocatalyst and gold-complex relay catalysis for the synthesis of spirooxindole derivatives.
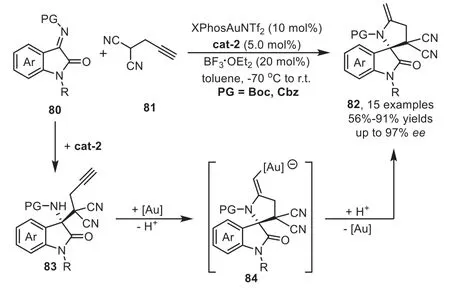
Soon after above report,Liu’s group has successfully developed a highly efficient one-pot sequential Mannich/hydroamination reaction of oxindole imines 80 with progargylated malononitrile 81 by a combined organocatalyst and XPhosAuNTf2relay catalysis for the expeditious construction of biologically active spirooxindole derivatives 82 in good overall yields and excellent enantioselectivities(Scheme 23)[89].During the optimization studies,they found that additive BF3·OEt2plays an important role in the goldcatalyzed hydroamination reaction to ensure the high catalytic activity by preventing the gold-complex from being poisoned by the quinidine derived organocatalyst cat-2.
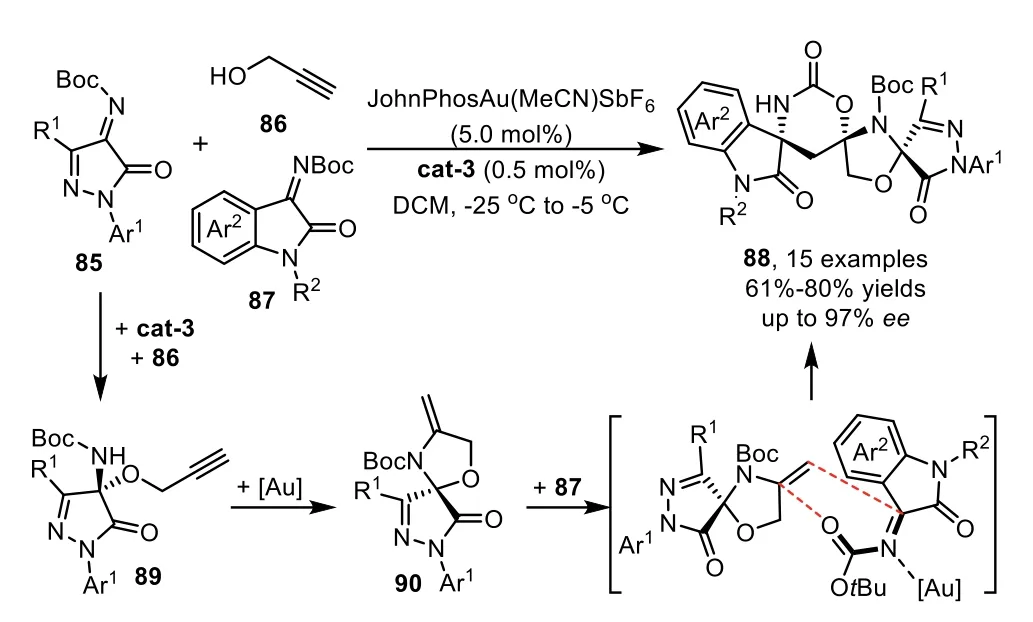
Recently,Li and co-workers have developed a urea derived bifunctional organocatalyst/gold-complex relay catalyzed cascade reaction for the direct assembly of chiral spirooxindoles from readily available materials[90].The cascade reaction comprises a highly efficient asymmetric 1,2-addition of propargyl alcohol 86 with imines 85 in the presence of chiral organocatalyst cat-3,and the generated chiral alkyne intermediates 89 occurs a triple cascade hydroamination/hetero-Diels-Alder(HDA)/debutylation sequence with the assistance of gold catalyst to deliver trispirocyclic spirooxindoles 88 in excellent stereoselectivity(Scheme 24).Notably,N-bociminooxindoles 87,usually used as the electrophiles in organic reactions,were found to be a good heterodiene for the HDA reaction with enamine 90.Moreover,the DFT calculations suggest that the key HDA reaction in the cascade process undergoes a concerted pathway.
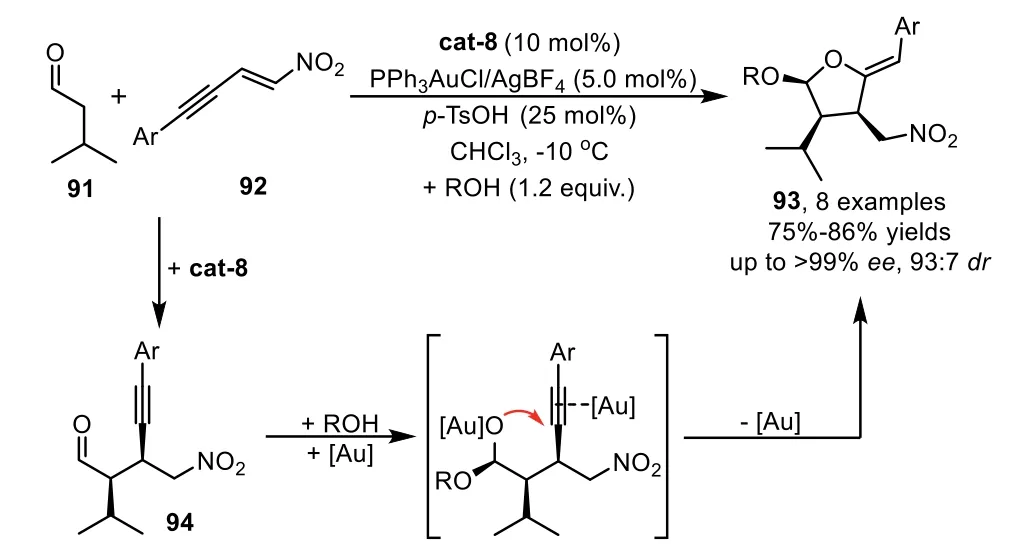
In addition to the asymmetric Mannich reaction,there are many other chiral organocatalysts that can promote different catalytic asymmetric methods for the preparation of enantioenriched alkynes,which could be subjected to the gold-catalyzed cyclization,leading to chiral cyclic frameworks with structural diversity.In 2009,Alexakis’group developed a one-pot cascade reaction involving an enantioselective amino oranocatalyst promoted Michael addition of aldehyde 91 with nitroenynes 92,and a subsequent gold-catalyzed tandem acetalization/cyclization of the corresponding adduct 94,leading to nitro-substituted tetrahydrofuranyl ethers 93 with high diastereo-and enantioselectivities(Scheme 25)[91].Moreover,they have demonstrated that amino organocatalysis cat-8 and gold-complex PPh3AuCl/AgBF4are compatible and complementary in this one-pot process,thereby the isolation yield of the products is higher than those obtainedviathe sequential procedures.
Scheme 25.Chiral amino organocatalyst and gold-complex relay catalysis for the synthesis of tetra-substituted tetrahydrofuran derivatives.
Scheme 26.Chiral thiourea derived organocatalyst and gold-complex relay catalysis for the synthesis of annulated indoles.
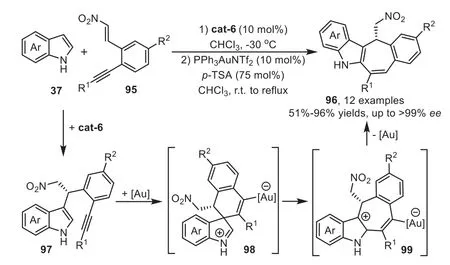
In 2011,Enders’group disclosed a novel methodology to access chiral tetracyclic indole derivatives by organocatalyst and gold-complex relay catalysis[92].The reaction starts from the first Friedel-Crafts type reaction of indoles 37 withortho-alkynesubstituted nitrostyrenes 95 in the presence of cat-6,which forms a tight transition state with these two substrates through multiple hydrogen-bonding interactions.Subsequently,the gold-activated adduct 97 allows the tethered indole to undergo the second Friedel-Crafts type reaction through a 6-endo-digfashion,forming a spirocyclic intermediate 98 which is confirmed by the control experiment.The intermediate 98 can rearrange to a sevenmembered ring form 99,followed by indole rearomatization and protodeauration steps to obtain the tetracyclic indole derivatives 96(Scheme 26).Three years later,the same group reported an analogous version of above cascade reaction with pyrroles 100 as the nucleophiles[93].The combination of a cinchona-alkaloid-derived primary amine cat-5 and a gold catalyst JohnPhosAuCl/AgNTf2gave excellent yields and enantioselectivity,which provides a convenient one-pot asymmetric synthesis of annulated pyrroles 102 from enones 101 based on the similar relay catalysis systemviaa sequential two Friedel-Crafts type cascade reaction(Scheme 27).
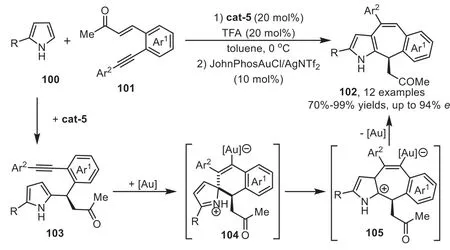
Scheme 27.Chiral quinidine derived organocatalyst and gold-complex relay catalysis for the synthesis of annulated pyrroles.
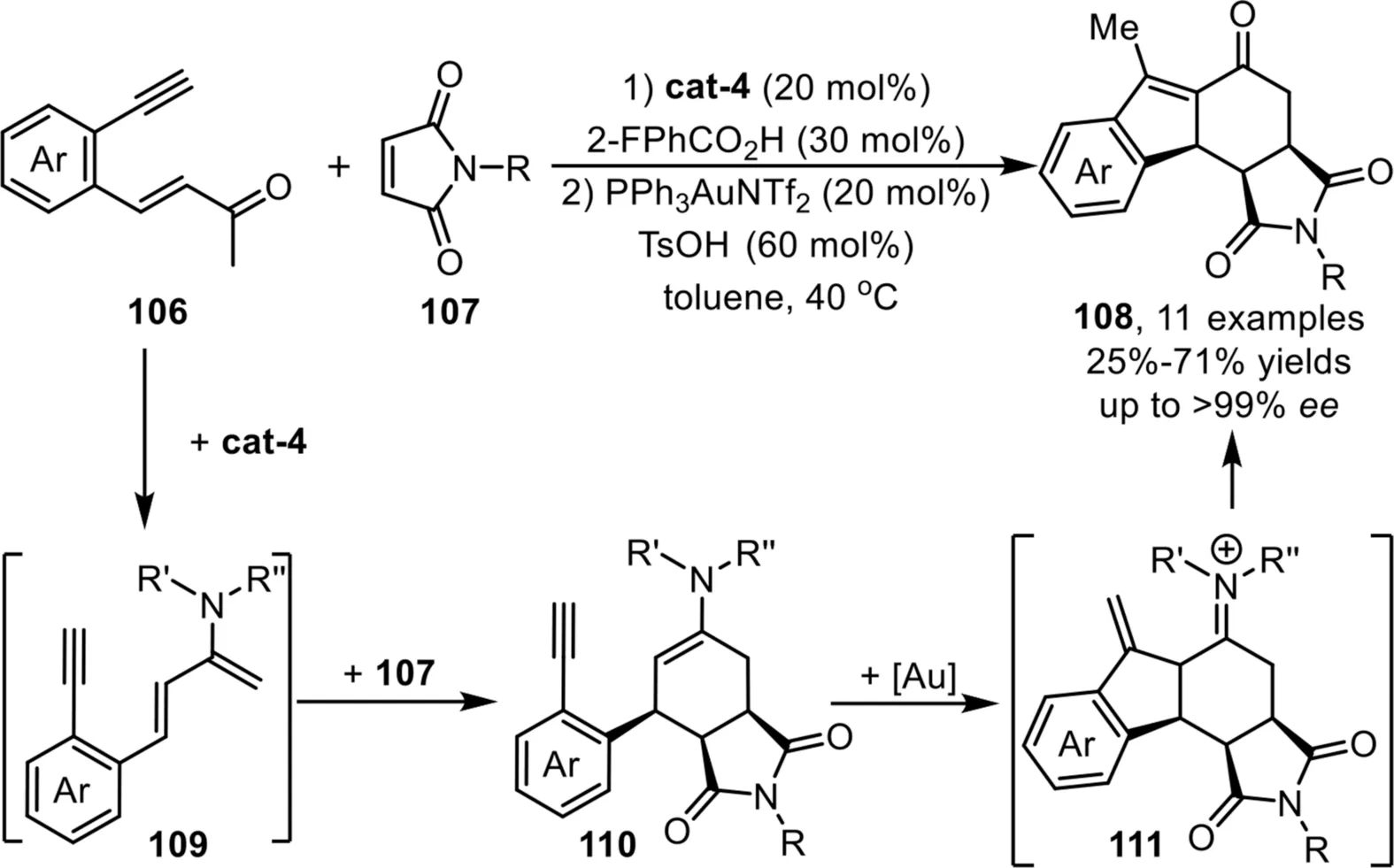
Scheme 28.Chiral quinidine derived and gold-complex relay catalysis for the synthesis of tetracyclic skeletons.
In 2014,Wu and coworkers have developed an efficient strategy for the enantioselective construction of[6,5,6]-carbocyclic compounds 108viaa one-pot sequential cyclization reaction of enones 106 with maleimides 107 under cinchona alkaloid-based primary amine and gold-complex relay catalysis[94].The reaction proceeds through a condensation reaction of enone 106 with chiral amine catalyst cat-4 to form an enamine intermediate 109,which would undergo a formal Diels-Alder reaction with maleimides 107,leading to the formation of an enamine intermediate 110.Subsequently,this intermediate would undergo an intramolecular carboannulation in the presence of PPh3AuNTf2to generate an iminium species 111,which could be readily transformed into the final ketone products 108 after hydrolysis,and the terminal carbon-carbon double bond migration driven by the formation of more thermodynamically stabilized internal form would occur simultaneously(Scheme 28).
Recently,Chan and co-workers developed an intramolecular cascade method for the enantioselective synthesis of 1,8-dihydroindeno[2,1-b]pyrroles in high to excellent yields and enantioselectivities under a sequential one-pot chiral Brønsted acid and gold-complex relay catalysis strategy[95].A tentative mechanism has been proposed that involves the formation of 1H-indene adduct 114viadehydrative Nazarov-type electrocyclization(DNE)ofβ-amino-1,4-enynols 112 in the presence of chiral Brønsted acid(S)-CPA-8.Subsequent activation of the alkyne moiety in the presence of the gold-complex would give the metal-associated adduct 115viaa 5-endo-dighydroamination,followed by protodeauration to deliver the tricyclic products 113(Scheme 29).
4.Gold-complex and chiral organocatalyst synergistic catalysis
In contrast to relay catalysis,the gold-complex and chiral organocatalyst synergistic catalysis involves a convergent assembly of gold associated intermediates,which derived from gold-catalyzed nucleophilic addition of alkynes,with chiral organocatalyst activated electrophiles,providing the chiral multifunctionalized products with two adjacent chiral centers(Scheme 30).In this context,three different synergistic catalysis models,including gold-complex and chiral Brønsted acid,gold-complex and chiral amine,and gold-complex and chiral squaramide-type catalyst synergistic catalysis will be discussed.
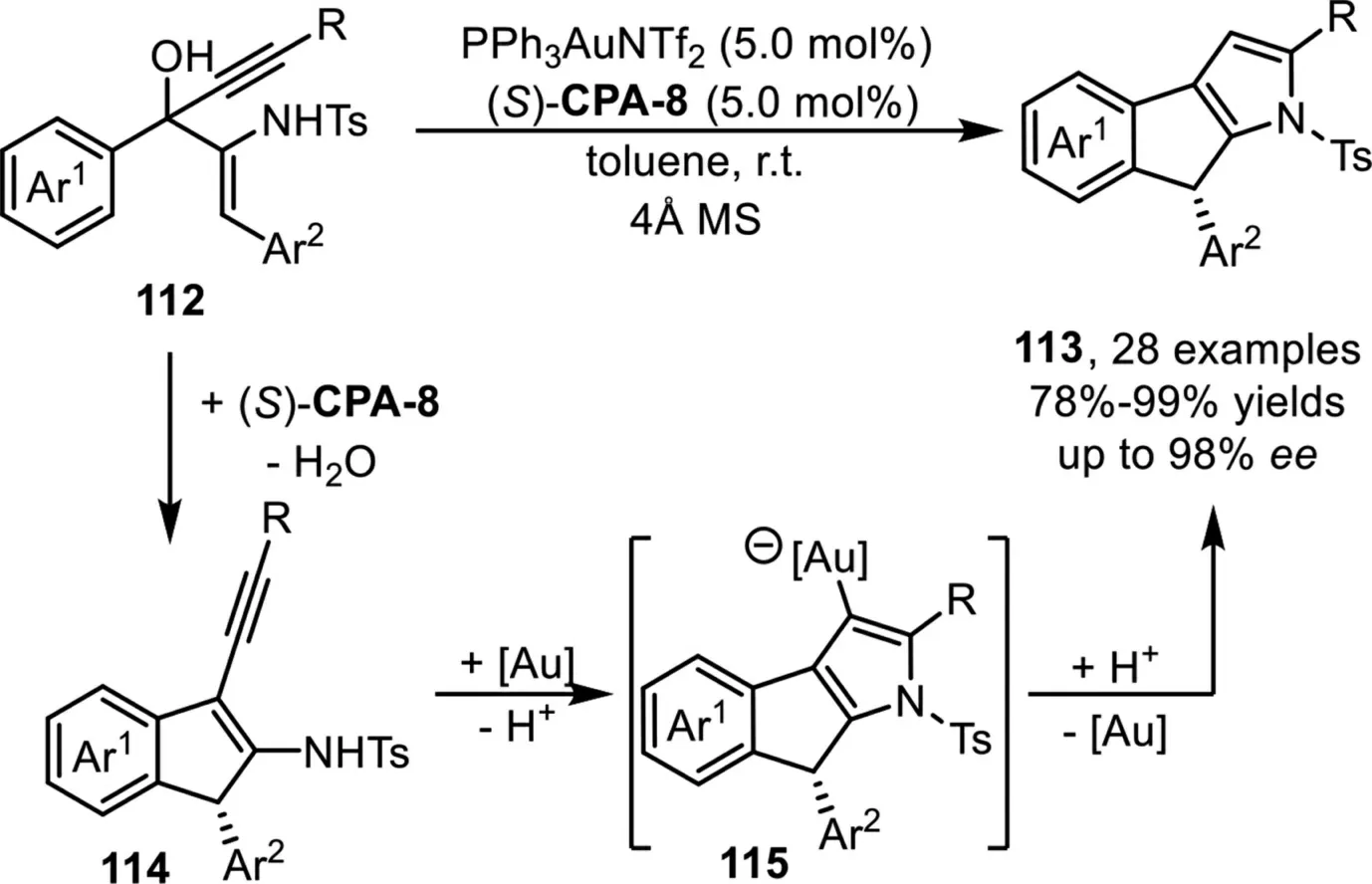
Scheme 29.Chiral Brønsted acid and gold-complex relay catalysis for the synthesis of 1,8-dihydroindeno[2,1-b]pyrroles.
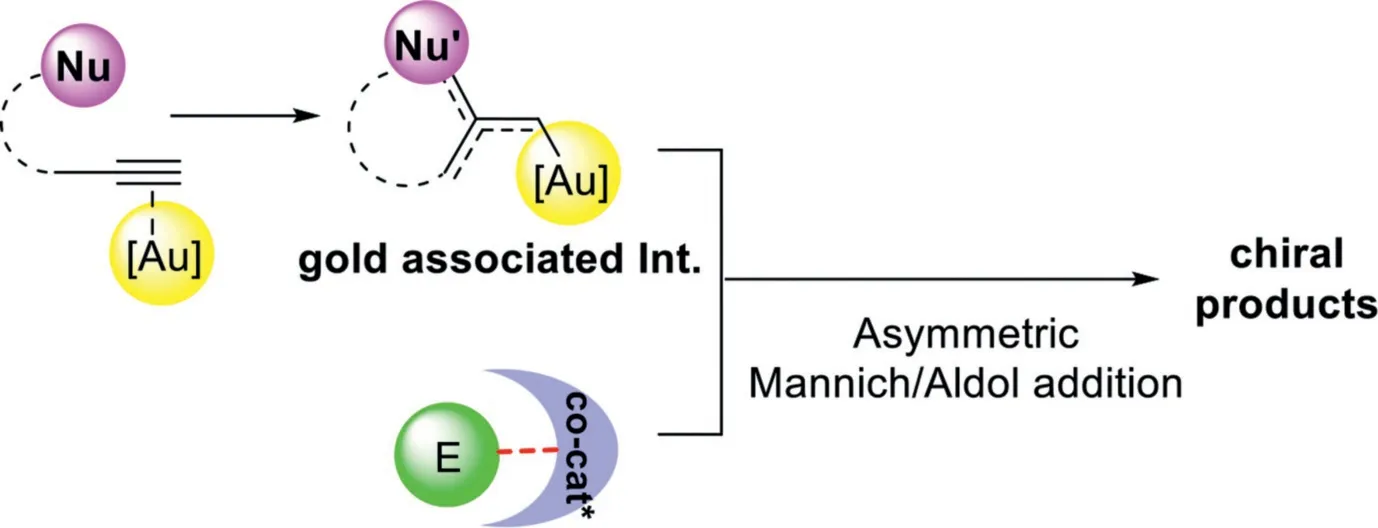
Scheme 30.Gold-complex and chiral organocatalyst synergistic catalysis.
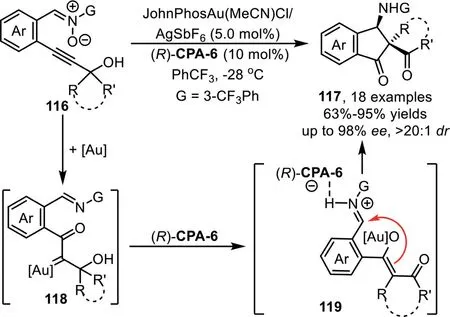
Scheme 31.Gold-complex and chiral phosphoric acid synergistic catalysis for the synthesis of chiralβ-amino spirocyclic diketones.
In 2013,J.Zhang and co-workers successfully developed a goldcomplex/chiral phosphoric acid cooperative catalysis system for the efficient synthesis of chiralβ-amino spirocyclic and quaternary diketone derivatives[96].The mechanism of this cascade reaction involves the initial formation of reactive goldα-oxo carbene intermediate 118 from nitrone-alkyne 116 in the presence of gold-complex.Subsequently,the carbene intermediate undergoes a rearrangement through a ring expansion process to give gold-associated enolate intermediate 119,which undergoes the Mannich-type reaction with the tethered imine species in an enantioselective fashion in the presence of chiral phosphoric acid(R)-CPA-6 to afford spirocyclic diketones 117(Scheme 31).
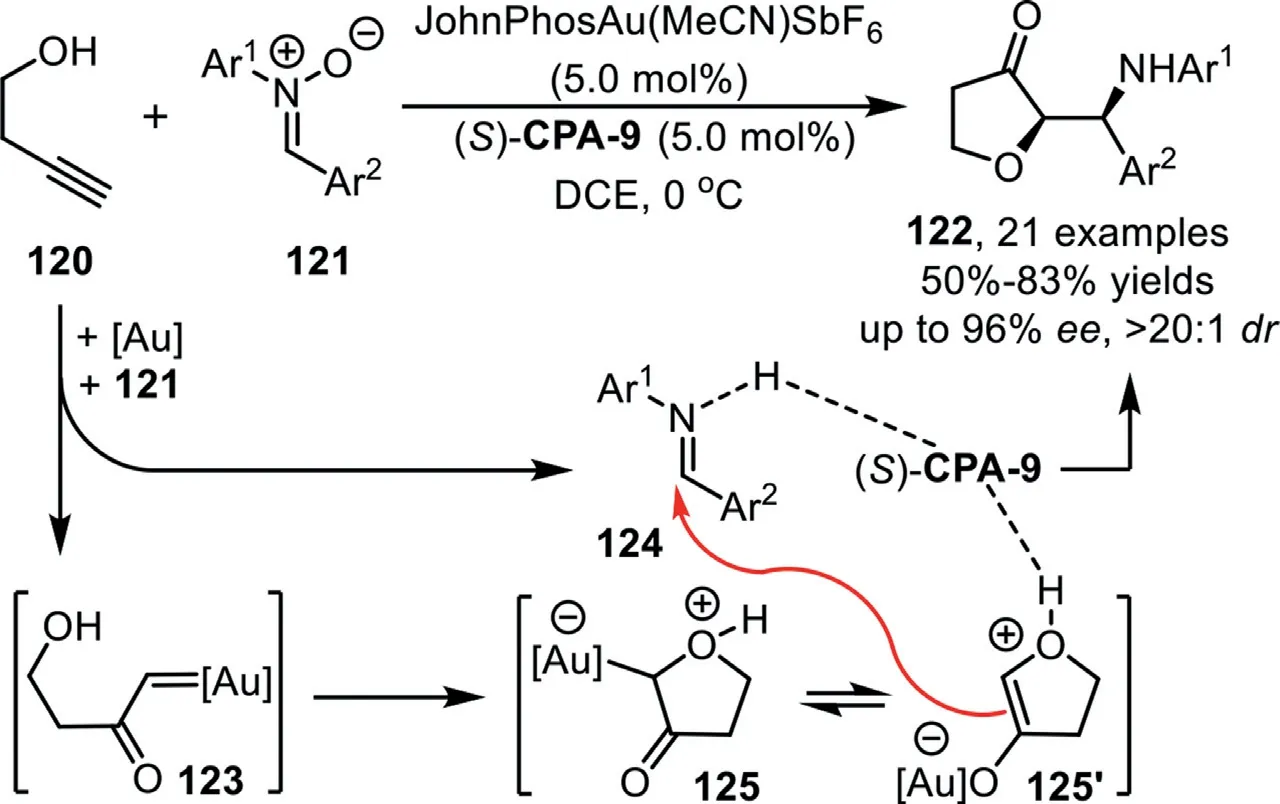
Scheme 32.Gold-complex and chiral phosphoric acid synergistic catalysis for the synthesis of dihydrofuran-3-one derivatives.
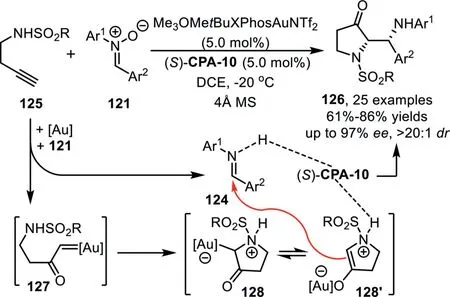
Scheme 33.Gold-complex and chiral phosphoric acid synergistic catalysis for the synthesis of pyrrolidin-3-one derivatives.
In 2018,Xu’s group reported a gold-complex/chiral phosphoric acid synergistic catalyzed Mannich-type reaction of 3-butynol 120 and nitrones 121 to afford dihydrofuran-3-one derivatives 122 in good yields with excellent diastereoselectivities and enantioselectivities[97].The reaction is initiated by gold-catalyzed alkyne oxidation to form the gold carbene species 123,followed by addition with the tethered hydroxy group to form oxonium ylide 125(or its enolate form 125′),which could be trapped through an enantioselective Mannich-type addition with the leaving fragment in the initial carbene formation step,imine,with the assistance of chiral phosphoric acid(S)-CPA-9(Scheme 32).Notably,this novel pattern of alkyne transformation involves a chemical bond cleavage,a fragment modification,and a reassembly process,providing an atom-and step-economic method for the asymmetric alkyne multi-functionalization with stable,inexpensive,and readily available materials.
Recently,the same group disclosed an efficient enantioselective method for the synthesis of chiral pyrrolidin-3-one derivatives under analogous synergistic catalysis conditions[98].In this reaction,homopropargyl amides 125,instead of 3-butynol,were used as carbene precursor,which form ammonium ylide 128(or its enolate form 128′)in the presence of gold-complex with nitrone as oxidant.Subsequently,an enantioselective Mannich-type addition with imine in the presence of chiral phosphoric acid(S)-CPA-10 led to the final products 126 in good to high yields with high to excellent enantioselectivities(Scheme 33).
At the same time,a highly enantioselective three-component reaction of terminal alkynes 2 with nitrones 121 and alcohols 129 has been reported by Xu’s group under gold-complex/chiral phosphoric acid catalysis conditions,which affordsα-alkoxy-β-aminoketones 130 with broad substrate generality in good to high yields and high to excellent enantioselectivities(Scheme 34)[99].Mechanistic studies suggest that both the gold-complex and chiral phosphoric acid(R)-CPA-11 contribute to the stereoselectivity determining step,providing a complementary protocol to the reported approach using diazoketones as carbene precursors,which generates diastereoisomeric products under achiral dirhodium complex and chiral phosphoric acid cooperative catalysis[100].
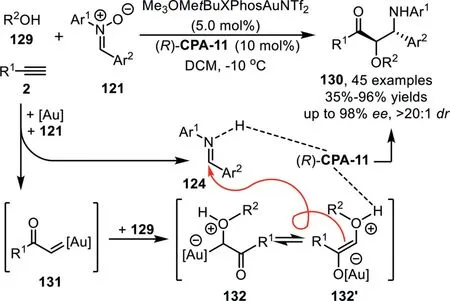
Scheme 34.Gold-complex and chiral phosphoric acid synergistic catalysis for the synthesis ofα-alkoxy-β-amino-ketones.

Scheme 35.Gold-complex and chiral amine synergistic catalysis for the synthesis of butanolide derivatives.
In addition to the chiral Brønsted acid,chiral amine and chiral squaramide-type catalysts are also compatible organocatalysts with gold-complex in one-pot reaction.Recently,Wang and coworkers developed a cascade synergistic catalysis strategy by combining chiral amino catalyst and gold-complex for the facile construction of the chiralγ,γ-disubstituted butenolides[101].Control experiments demonstrate the synergistic effect of this binary catalytic system.The amine catalyst cat-8 not only serves as a base to facilitate the gold-catalyzed(or silver)cyclization of alkynoic acids 133viathe generation of more nucleophilic anionic form species;but also assists the cyclized adduct 136 to form its gold-associated enolate species 136′.Subsequently,the chiral amine mediated conjugate addition of this intermediate with activated aldehyde 134 led to the final products 135 and regenerated the catalyst(Scheme 35).Notably,this method could be applied to the synthesis of a key intermediate towards natural product(−)-aromdendranediol.
In 2021,Xu and co-workers reported the first example of asymmetric allylation reaction of isatins and isatin-derived ketimines withN-propargylamides that enabled by gold-complex and chiral squaramide-type catalyst,which offers access to chiral 2,5-disubsituted alkylideneoxazolines generally in an optically pure form[102].This reaction is initiated through a gold-catalyzed 5-exo-digcyclization ofN-propargylamide 138 to deliver the key(E)-vinylgold intermediate 141,followed by a H-shift driving by the aromatization under mild basic conditions,leading to the other key alkylgold species 142,which has been confirmed and characterized by X-ray crystallographic analysis for the first time.The final asymmetric allylation process of this allylgold species through a formal hetero-ene reaction is realized by the bifunctional chiral organocatalyst cat-7,forming the final products 140 and regenerate the catalysts(Scheme 36).
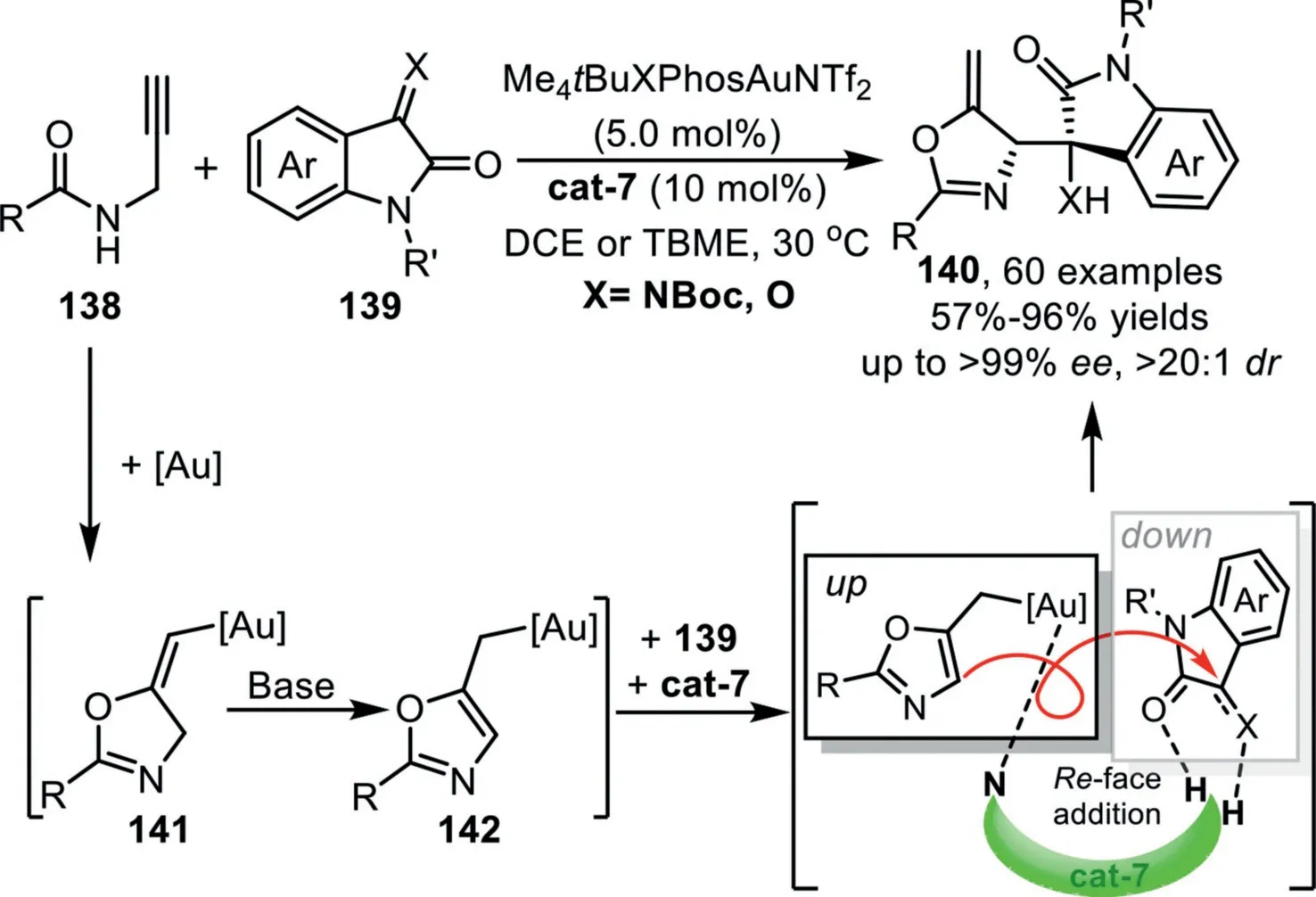
Scheme 36.Gold-complex and chiral squaramide-type synergistic catalysis for the synthesis of 2,5-disubsituted alkylideneoxazolines.
5.Summary and outlook
In this review article,the gold-complex and chiral organocatalyst cooperative catalysis in catalytic asymmetric alkyne functionalization has been comprehensively summarized and divided by different catalytic models,including relay catalysis and synergistic catalysis.In comparison to the other types of asymmetric catalysis methods in alkyne transformations,cooperative catalysis has demonstrated advantages in many aspects,especially enabling novel asymmetric reactions for the alkyne multi-functionalizations.Although progresses have been made in recent decades as listed in this review,limitations remain in this area:1)the types of goldcomplex compatibility with organocatalysts are still restricted,especially for the amino organocatalysts,additives or complicated condition optimization are inescapable;2)most of the cascade reactions occurs intramolecularly with prefunctionalized alkynes,robust catalysts or/and catalytic conditions are continuingly needed for the effective and selective activation of readily available alkyne materials;3)synthetic applications for asymmetric alkyne multifunctionalization and synthesis of complex bioactive molecules are still under exploring.We anticipate that this review will not only give the generalization and summary of cooperative catalysis models in asymmetric alkyne transformations,but also shed light on the prospect of the design and development of more sophisticated cooperative asymmetric catalysis methods by exploration of versatile gold compatible chiral organocatalysts,and by using of goldchiral Lewis acid cooperative catalysis,which is still limited few examples but with prospective potentials.Novel synthetic methods and applications could be envisioned that enabled by these cooperative catalysis strategies through successful interception of these either confirmed or proposed prochiral gold-associated intermediates.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Support for this research from the National Natural Science Foundation of China(Nos.21971262,81702255),National Postdoctoral Program for Innovative Talents(No.BX20190399),Guangdong Provincial Key Laboratory of Chiral Molecule and Drug Discovery(No.2019B030301005),and The Program for Guangdong Introducing Innovative and Entrepreneurial Teams(No.2016ZT06Y337)is greatly acknowledged.
杂志排行

Chinese Chemical Letters的其它文章
- Diverse strategic approaches en route to Taxol total synthesis
- Unmodified methodologies in target discovery for small molecule drugs:A rising star
- Recent advances in single-crystalline two-dimensional polymers:Synthesis,characterization and challenges
- Environmental applications of graphene oxide composite membranes
- Recent advances in the application of metal organic frameworks using in advanced oxidation progresses for pollutants degradation
- Recent progress on two-dimensional materials confining single atoms for CO2 photoreduction