PPARs激动剂对糖尿病伤口的促愈合机制及研究进展
2023-01-16鲍蕾蕾李晓龙蒋会荣台宗光朱全刚信如娟
黄 皓,鲍蕾蕾,李晓龙,蒋会荣,台宗光,朱全刚,信如娟*
0 引言
糖尿病是一种由于胰岛素分泌和(或)利用缺陷引起的代谢性疾病,发病率高,增长快,预计2045年将有7.83亿成年人受到该疾病的影响[1-2]。糖尿病伤口难以愈合作为糖尿病常见的症状之一,涉及多学科、多专业,病理复杂,处理棘手,是多种不利因素共同作用的结果。糖尿病伤口的医疗支出巨大,而且会给患者带来身体和精神上的双重打击,严重者可以导致患者抑郁、截肢甚至死亡[3]。过氧化物酶体增殖物激活受体(Peroxisome proliferator-activated receptors,PPARs)属于核激素受体家族[4],有3种不同的亚型:PPARα、PPARγ、PPARβ/δ,在人体内分布广泛,不仅可以用于治疗胰岛素抵抗,还可以通过调节脂肪酸稳态、抑制过度炎症、改善创面的增殖和重塑等作用促进伤口愈合。近年来,PPARs激动剂作为治疗伤口难愈的潜在策略已经受到越来越多的关注[5-6],但PPARs激动剂促进糖尿病慢性伤口愈合的作用机制尚未得到全面、系统地说明。本文阐述了PPARs激动剂在糖尿病伤口愈合中的作用机制和研究进展,为糖尿病伤口愈合提供了新的治疗思路。
1 PPARs激动剂在糖尿病伤口愈合发病机制中的作用
伤口愈合需要经过4个连续而重叠的阶段:止血、炎症、增殖和重塑[7]。在高糖环境下,脂质代谢异常、免疫细胞和皮肤细胞功能受损、血管病变等都会影响伤口的愈合。PPARs激动剂通过激活PPARs参与伤口愈合,并在各个阶段发挥作用。见图1。
1.1 调节脂肪酸稳态 胰岛功能受损,使得糖尿病患者体内的脂质代谢酶活性下降,细胞储存脂肪的功能受损[8];多饮、多食的特征使得患者脂质的日常摄入量增加,两者共同造成患者血液中的游离脂肪酸(Free fatty acid,FFA)增多,脂肪沉积增加,导致患者的胰岛素抵抗加重,血管硬化,进一步加重了患者高血糖的症状,胰岛功能下降和脂肪酸稳态失衡的相互作用可导致伤口的微环境渐进式螺旋恶化,从而抑制了伤口的愈合。PPARs通过调节脂质细胞的代谢分化和减轻胰岛素抵抗,起到治疗糖尿病慢性伤口的作用,促进了伤口的愈合[9]。见图2。

图1 PPARs在糖尿病慢性伤口愈合中的作用注:红色箭头代表促进或增加,绿色箭头代表抑制或减少,红色字体代表过量。PPARs:过氧化物酶体增殖物激活受体;NF-κB:核因子κB;TNF-α:肿瘤坏死因子-α;COX-2:环氧合酶-2;iNOS:诱导型一氧化氮合酶;eNOS:内皮型一氧化氮合酶;MMP-9:金属蛋白酶-9;PI3K:磷脂酰肌醇3-激酶;TAK1:TGF-β激活激酶1;AP1:核转录激活蛋白1;TGF-β:生长转化因子-β;VEGF:血管内皮生长因子;FGFs:成纤维细胞生长因子;IL-1Ra:重组白介素-1受体拮抗剂;PDK1:3-磷酸肌苷依赖激酶-1;ILK:整合素连接激酶;PKB:蛋白激酶B;FKHR:叉头转录因子;GSK-3β:糖元合成酶激酶3β
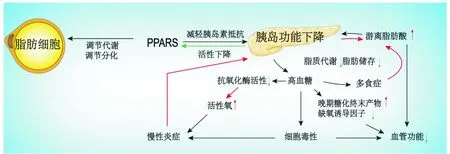
图2 胰岛功能下降和脂肪酸稳态失衡的相互作用共同抑制糖尿病伤口愈合注:红色箭头代表促进或增加,绿色箭头代表抑制或减少。PPARs:过氧化物酶体增殖物激活受体
脂肪细胞释放的炎性介质(包括细胞因子和生物活性脂质)影响了免疫细胞和皮肤细胞的功能,脂肪细胞作为配体或通过释放炎性介质激活PPARs[10]。PPARs被激活后,影响脂肪细胞的代谢和分化,调节人体脂肪酸稳态[11]。研究表明,PPARα在细胞水平的增加,改善了FFA的代谢和β氧化,降低了胰岛素抵抗和脂肪酸代谢紊乱[12]。PPARα和PPARγ被激活后,可减少油酸处理后的巨噬细胞中的脂质积累,降低由于脂肪沉积所导致的血管疾病的风险[13]。PPARβ/δ的激活能够提高血浆中高密度脂蛋白胆固醇(High density lipoprotein,HDL)水平,并降低极低密度脂蛋白胆固醇(Very low density lipoprotein,VLDL)水平。综上所述,PPARs的激活缓解了胰岛素抵抗和脂肪沉积对伤口愈合的不利作用,促进了伤口的愈合。
1.2 抑制过度炎症 伤口愈合的炎症阶段,受损的皮肤细胞持续释放生长因子,促进免疫细胞在伤口的聚集,以保证损伤部位炎症反应的正常运行,进而抵抗病原体的入侵,促进伤口的愈合。
在高血糖环境中,抗氧化酶的活性降低,晚期糖基化终末产物(Advanced glycation end products,AGEs)增加,使伤口长期处于氧化应激状态,导致创面缺氧[14]。炎症阶段的加剧和延长,使得巨噬细胞无法从促炎型向抑炎型转化,限制了巨噬细胞在伤口增殖、重塑阶段的作用[15-16]。同时,持续的慢性炎症,使得炎症过程中的负反馈因子异常磷酸化[17],干扰了与胰岛素相关的信号通路,加重了胰岛素抵抗,抑制了胰岛素的抗炎、调节代谢的能力[18],胰岛素的减少也会导致PPARs的活性下降[19]。
PPARα参与调控伤口早期的炎症反应,可促进巨噬细胞、中性粒细胞向伤口募集,在伤口愈合的早期阶段发挥重要作用[20]。PPARα通过调节核因子-κB抑制剂(Inhibitor kappa B kinase,IKK-B)的基因表达,干预核因子κB(Nuclear factor kappa B,NF-κB)通路的促炎作用,并诱导激发PPARγ在炎症期间的抑炎作用[21]。
巨噬细胞吞噬凋亡细胞后,凋亡细胞在巨噬细胞内裂解后释放的胆固醇和脂肪酸是PPARγ的激动剂,激活后的PPARγ可以抑制核受体阻遏物(Nuclear receptor co-repressor,NCoR)与NF- κB的位点分离,进而抑制 NF-κB信号通路的激活[22],促进巨噬细胞从促炎型到抑炎型的转化,使伤口愈合从炎症阶段正常过渡至增殖、重塑阶段。
皮肤创伤的炎症反应上调了PPARβ/δ的表达[23],PPARβ/δ通过抑制NF-κB信号通路和巨噬细胞中促炎因子,如诱导型一氧化氮合酶(Inducible nitric oxide synthase,iNOS)、环氧合酶-2(Cyclooxygenase-2,COX2)、肿瘤坏死因子(Tumor necrosis factor-α,TNF-α),在炎症环境中发挥促进伤口愈合的效果。
1.3 改善创面的增殖和重塑
1.3.1 调节皮肤细胞的增殖和迁移 高糖环境下的细胞毒性,抑制了成纤维细胞的增殖和分化能力[24],成纤维细胞分泌转化生长因子-β(Transforming growth factor-β,TGF-β)的功能受损,胶原蛋白合成减少,阻碍了伤口的愈合,同时高糖的慢性炎症环境,使得基质金属蛋白酶(Matrix metalloproteinase,MMP)过度表达,导致胶原蛋白降解过度,角质细胞的迁移不受控制,伤口难以愈合。
炎性介质和其增强的信号通路共同导致了PPARβ/δ的激活[25],上调了整合素连接激酶(Integrin-linked kinase,ILK)和3-磷酸肌苷依赖激酶-1(3-phosphoinositide-dependent protein kinase-1,PDK1)的表达,进而激活下游信号通路发挥调控角质形成细胞存活、黏附,迁移功能的作用。研究显示,敲除PPARβ/δ,会使小鼠伤口的愈合时间延长2~3 d[26]。
PPARβ/δ也是调节成纤维细胞和角质形成细胞增殖平衡的重要受体[27]。角质细胞通过分泌白细胞介素-1(Interleukin-1,IL-1)与成纤维白细胞介素-1受体结合,通过成纤维细胞的TGF-β激活激酶1(TGF-β-activated kinase 1,TAK1)/c-Jun/核转录激活蛋白1(Activator protein-1,AP-1)信号通路释放生长因子,促进自身和成纤维细胞的增殖。PPARβ/δ的激活可促进成纤维细胞中重组白介素-1受体拮抗剂(Interleukin-1 receptor antagonist,IL-1Ra)的生成,从而抑制IL-1在角质细胞中的促增殖作用,防止角质形成细胞过度增殖。
1.3.2 促进血管的修复 缺血及缺血导致的供氧不足、营养不足是造成糖尿病皮肤伤口难愈的主要因素,血管损伤也是糖尿病皮肤溃疡患者最常见的临床症状。通常情况下,内皮型一氧化氮合酶(Endothelial nitric oxide synthase,eNOS)参与了血管内皮生长因子和成纤维生长因子(Fibroblast growth factor,FGF)促进细胞增殖的过程,通过释放一氧化氮(Nitric oxide,NO),促进了血管的生长和通透性,保证了营养和氧气的供应;在高血糖的情况下,高浓度的葡萄糖和AGEs消耗了NO,并且抑制eNOS的活性,NO的合成减少,使得生长因子无法发挥作用[28],造成血管损伤难以修复。糖尿病患者血液中高浓度葡萄糖与肢体微血管壁中的胶原形成异常胶原蛋白沉淀,从而导致血管弹性丧失,微循环变差。高糖也可降低缺氧诱导因子1α(Hypoxia-inducible factor 1-α,HIF-1α)的活性,造成许多重要蛋白质的非酶糖化,导致细胞和细胞外基质功能异常,从而抑制糖尿病伤口血管的修复[29]。
有研究表明,PPARβ/δ激动剂可以调节血管内皮生长因子、成纤维生长因子、血栓素和内皮抑素等的表达,也可以MMP-9依赖的方式诱导胰岛素样生长因子(Insulin-like growth factor-1,IGF-1)受体磷酸化,这些都有利于血管的修复。PPARβ/δ激动剂还通过激活LKB1/AMP依赖蛋白激酶[Adenosine 5′-monophosphate (AMP)-activated protein kinase,AMPK]/eNOS信号通路增加eNOS的表达,使糖尿病小鼠主动脉内皮依赖性舒张正常化,起到保护血管的作用[30]。PPARγ激动剂罗格列酮可上调HIF-1α、HIF-2α的mRNA表达水平,调节血管内皮生长因子、血管紧张素的表达,增强血管内皮细胞的黏附、增殖和迁移能力,有利于血管的形成[31]。
2 PPARs激动剂的研究进展
2.1 PPARs激动剂在糖尿病伤口愈合中的临床研究 Bonora等[32]在随机临床对照试验中证明:非诺贝特(PPARα激动剂)可以通过增加体内的循环造血干细胞和祖细胞(Circulating haematopoietic stem/progenitor cells,HSPCs)维护血管功能。一项关于罗格列酮的临床研究表明,男性糖尿病患者在服用罗格列酮(PPARγ激动剂)1年后,患者脂肪细胞中脂肪酸运输、合成和储存的基因转录增加,血液中的FFA减少,罗格列酮还通过NO信号通路减少了血管炎症,促进了内皮细胞功能的修复,有利于损伤血管的再生[33]。
2.2 PPARs激动剂的研发方向和研究进展 PPARs激动剂目前有2个研发方向,一是通过改进剂型降低PPARs过度激活带来的不良反应:吡格列酮改进为局部缓释制剂后,具有生物利用度高、全身副作用少等优点,可在伤口愈合的炎症和增殖阶段发挥重要作用[5];PPARs泛激动剂IVA337局部注射具有抗炎、抗纤维化的作用,且不会延缓伤口的愈合[34],可用于糖尿病伤口的治疗;二是寻找安全、平衡的可激活多种PPARs的药物:PPARs泛激动剂MHY2013改善了小鼠因肥胖引起的胰岛素抵抗、血脂异常和肝脂肪变性[35],药物毒性低,具有促进糖尿病伤口愈合的潜力;GQ-11作为PPARs双激动剂(PPARα、PPARγ),在体内外给药均可以促进抗炎因子和促愈合因子的表达,加速了糖尿病小鼠创面上皮细胞的形成和胶原沉积[16];全球首个上市的PPARs泛激动剂Chiglitazar在临床中可用于降糖和改善胰岛素抵抗[36],能激活PPARα、PPARβ/δ、PPARγ 3种亚型,其在促进糖尿病伤口愈合中的潜力有待进一步探索。
3 展望
综上所述,PPARs激动剂可通过调节脂肪酸稳态、抑制过度炎症、改善创面的增殖和重塑等作用促进伤口的愈合,为伤口愈合提供治疗策略。然而,目前关于PPARs激动剂的报道不能支撑推荐这些药物作为伤口愈合的一线治疗方式。未来更多的体外和体内研究可能会揭示PPARs在伤口愈合中的作用,并评估其在伤口愈合的功效和安全性。
